
journalsclub-blog
"JOURNALS CLUB & CO."
DR. PORAS PATEL, DIRECTOR, AHMEDABAD, GUJARAT, INDIA
53 posts
Don't wanna be here? Send us removal request.
Last Seen Blogs

alstruprobertson94
The Life of Kelleher 576

yotakafas
inside the head of an artist 🍉

pixiie-boy
parker
yotakafas
inside the head of an artist 🍉

histrionicscribbler
wait a minute... who ARE you
Text
Heat Shock Proteins, a short review
INTRODUCTION
Stressful conditions trigger certain defence mechanisms, including those at molecular levels. This was first seen in Drosophilia and was reported in 1974.1 Heat Shock Proteins (HSPs), also known as Stress-induced Proteins or Stress Proteins, are one such class of proteins that are produced in the body in response to stress, under the control of Heat Shock Factors (HSFs), although some are constitutively expressed. The stress may be heat, cold, UV radiation, infections, inflammation, heavy metal exposure or else. HSPs are produced by all organisms and are ubiquitously present. The primary involvement of these proteins is in the folding and stabilization of other proteins, and thus they play an intimate role in the
aggregation of various other proteins.2 Besides action on protein folding, these HSPs also possess pro- and anti-apoptotic properties, making them suitable targets for drug development. The HSP families are classified according to their molecular weight.3 Table 1 describes in brief the classification as well as a few functions of these proteins.
In the recent past, various drug have been developed which act in line or against the HSPs but a still in their infancy. Besides drugs, the HSPs are also employed as diagnostic tools in various cancers. These are referenced in Table 2.
The flip side is the new set of adverse effects which are seen with these class of drugs. In patients with H.pylori infection which is implicated in the development of gastric carcinoma, it was observed that HSPs contributed to the progression of H. pylori-associated gastric carcinogenesis as well as led to the aggravation of gastric inflammation.33
Table 1: Functions of Heat Shock Proteins
Family
Function
HSP90
(constitutive, induced)4 - 8
- Regulatory interactions with signalling proteins
- Protein synthesis, folding and degradation
- Stabilization of misfolded proteins
- Binding of estrogen, progesterone, androgen, and aldosterone 5
- Delivery of antigens to APCs 6
- Cancer cells: enhances growth, supresses senescence, provides resistance to stress induced apoptosis. 7
- Cardioprotective: binds to NO synthase and Guanylate cyclase, cause vascular relaxation 8
HSP70
(constitutive)
6, 9 - 12
- Protein folding, membrane transport of proteins 9
- Anti-apoptotic 10
- Delivery of antigens to APCs 6
- In sympathetic neurons: 11
• HSP 72 – inhibits degradation of Tau protein, heat shock inducible
• HSC 70 - promotes degradation of Tau protein
Low levels – associated with insulin resistance 12
HSP60
(constitutive)
- In the mitochondria, replication and transcription of DNA, pro-survival. 13
- In the cytosol, complexes and inhibits maturation and activation of Caspase 3 – Anti apoptotic 14
- At the surface and extracellularly, stimulates immune response 15
HSP40
- Protein folding, co-chaperon for HSP70 16
- HSP40-70 complex – modulate accumulation of polyglutamine proteins 17
HSP27 (β1) (induced)
- Anti-apoptotic, prevents proteolysis by inhibiting liberation of cytochrome c from mitochondria18
Small HSPs
- Stabilization of misfolded proteins19
Table 2: Drugs acting via HSPs
Family
Drug
Disease
Against HSP90
Geldanamycin
(derivative, 17-allylamine,17-demethoxigeldanamycin)
Malaria20
Huntington’s disease21
Cacncers22,23
Efungumab
Invasive Candidiasis24
Against HSP70
Triptolide
Pancreatic cancer25
Mesothelioma26
Methylene blue (inhibits ATPase activity of HSP72)
Alzheimer’s disease27
Pro-HSP60
Bortezomib28
Malignancies, increases expression of HSP60 on malignant cells and thus enhances immune response against tumour cells
Against HSP40
Quercetin (inhibits HSP 40 and 27)
Parkinson’s disease29
Cancer30
Against HSP27
Apatorsen (antisense oligonucleotide)
Cancer31
Diagnostic tool32
Increased levels - Renal injury and fibrosis, Cancers of breast, lung, liver, prostate, rectal, osteosarcoma, leukaemia, cerebral and cardiac ischemia
Reduced levels – oesophageal cancer
Anti-HSP27 IgA – Gynaecological malignancies
Against HSP70
Triptolide
Pancreatic cancer25
Mesothelioma26
Methylene blue (inhibits ATPase activity of HSP72)
Alzheimer’s disease27
Pro-HSP60
Bortezomib28
Malignancies, increases expression of HSP60 on malignant cells and thus enhances immune response against tumour cells
Against HSP40
Quercetin (inhibits HSP 40 and 27)
Parkinson’s disease29
Cancer30
Against HSP27
Apatorsen (antisense oligonucleotide)
Cancer31
Diagnostic tool32
Increased levels - Renal injury and fibrosis, Cancers of breast, lung, liver, prostate, rectal, osteosarcoma, leukaemia, cerebral and cardiac ischemia
Reduced levels – oesophageal cancer
Anti-HSP27 IgA – Gynaecological malignancies
Autoimmune disease: Since these are highly conserved in nature, they are the initiators as well as the targets of autoimmune attack. Molecular mimicry and cross presentation of antigens are the phenomena of their involvement in autoimmunity. Their roles have been implicated in atherosclerosis, uveitis, lupus and Behcet’s disease. 34
Atherosclerosis: Risk factors for atherosclerosis including infection, oxidative stress, biomechanical stress, all lead to the overproduction of HSPs through the activation of heat shock transcription factor 1 which may lead to worsening of atherosclerosis.35
The anti-apoptotic property may lead to a poor prognosis and resistance to therapy in cancer which the anti-apoptotic activity may be therapeutically advantageous.36
Insomnia or sleep deprivation can lead to an increased level of HSPs acting as a neuroprotective response, emphasizing on the role of adequate sleep in disease prevention.37
CONCLUSION
Harms and benefits are two sides of the same coin, as is the case with heat shock proteins. Despite their presence ubiquitously, a small rise or fall in their levels can have a different specific new set of adverse implications. However, despite the availability of information, further research in needed in order to develop newer drugs which may prove beneficial in the treatment of difficult, incurable diseases.
REFERENCES
Schlesinger, M. J. (1990). Heat shock proteins. The Journal of Biological Chemistry, 265(21), 12111–12114. PMid:2197269
Lindquist, S., & Craig, E. A. (1988). The heat-shock proteins. Annual review of
genetics, 22(1), 631-677. https://doi.org/10.1146/annurev.ge.22.120188.003215, PMid:2853609
Wirth, D., Gustin, P., Drion, P., Dessy-Doize, C., & Christians, E. S. (2002). Heat shock proteins. I: Classification and roles in pathological processes. In Annales de Médecine Vétérinaire (Vol. 146, No. 4, pp. 201-216). Annales Medecine Veterinaire.
Jackson, S. E. (2012). Hsp90: structure and function. In Molecular chaperones(pp. 155-240). Springer, Berlin, Heidelberg. https://doi.org/10.1007/128_2012_356,
PMid:22955504
Joab, I., Radanyi, C., Renoir, M., Buchou, T., Catelli, M. G., Binart, N., ... & Baulieu, E. E. (1984). Common non-hormone binding component in non-transformed chick oviduct receptors of four steroid hormones. Nature, 308(5962), 850. https://doi.org/10.1038/308850a0,
PMid:6201744
Gaston, J. S. H. (2002). Heat Shock Proteins and Innate immunity. Clin Exp Immunol, 127(1), 1–3. https://doi.org/10.1046/j.1365-2249.2002.01759.x,
https://doi.org/10.1111/j.1365-2249.1990.tb05394.x, PMid:11882025, PMCid:PMC1906278
Workman, P., Burrows, F., Neckers, L., & Rosen, N. (2007). Drugging the cancer chaperone HSP90: combinatorial therapeutic exploitation of oncogene addiction and tumor stress. Ann N Y Acad Sci, 1113, 202–216. https://doi.org/10.1196/annals.1391.012,
PMid:17513464
Antonova, G., Lichtenbeld, H., Xia, T., Chatterjee, A., Dimitropoulou, C., & Catravas J. D. (2007). Functional significance of hsp90 complexes with NOS and sGC in endothelial cells. Clin Hemorheol Microcirc, 37(1-2), 19-35.
Hartl, F.U. (1996). Molecular chaperones in cellular protein folding. Nature, 381, 571-579. https://doi.org/10.1038/381571a0,
PMid:8637592
Gehrmann, M., Radons, J., Molls, M., & Multhoff, G. (2008). The therapeutic implications of clinically applied modifiers of heat shock protein 70 (Hsp70) expression by tumor cells. Cell Stress Chaperones, 13(1), 1-10. https://doi.org/10.1007/s12192-007-0006-0, PMid:18347936, PMCid:PMC2666213
Jinwal, U. K., Akoury, E., Abisambra, J. F., O'Leary, J. C., Thompson, A.D., Blair, L. J., et al. (2013). Imbalance of Hsp70 family variants fosters tau accumulation. FASEB J, 27(4), 1450-1459. https://doi.org/10.1096/fj.12-220889, PMid:23271055, PMCid:PMC3606536
Chichester, L., Wylie, A. T., Craft, S., & Kavanagh, K. (2015). Muscle heat shock protein 70 predicts insulin resistance with aging. J Gerontol A Biol Sci Med Sci, 70(2), 155-62. https://doi.org/10.1093/gerona/glu015,
PMid:24532784, PMCid:PMC4311181
Kaufman, B. A., Kolesar, J. E., Perlman, P. S., & Butow, R. A. (2003). A function for the mitochondrial chaperonin Hsp60 in the structure and transmission of mitochondrial DNA nucleoids in Saccharomyces cerevisiae. J Cell Biol, 163, 457-461. https://doi.org/10.1083/jcb.200306132,
PMid:14597775, PMCid:PMC2173642
Xanthoudakis, S., Roy, S., Rasper, D., Hennessey, T., Aubin, Y., Cassady, R., et al. (1999). Hsp60 accelerates the maturation of pro-caspase-3 by upstream activator proteases during apoptosis. EMBO J, 18(18), 2049-2056. https://doi.org/10.1093/emboj/18.8.2049,
PMid:10205159, PMCid:PMC1171289
Pockley, A. G., Muthana, M., & Calderwood, S. K. (2008). The dual immunoregulatory roles of stress proteins. Trends Biochem Sci, 33, 71-79. https://doi.org/10.1016/j.tibs.2007.10.005,
PMid:18182297
Castanie-Cornet, M. P., Bruel, N., & Genevaux, P. (2014). Chaperone networking facilitates protein targeting to the bacterial cytoplasmic membrane. Biochim Biophys. Acta, 1843, 1442-1456. https://doi.org/10.1016/j.bbamcr.2013.11.007,
PMid:24269840
Wacker, J. L., Zareie, M. H., Fong, H., Sarikaya, M., & Muchowski, P. J. (2004). Hsp70 and Hsp40 attenuate formation of spherical and annular polyglutamine oligomers by partitioning monomer. Nature Structural & Molecular Biology, 11(12), 1215-1222. https://doi.org/10.1038/nsmb860,
PMid:15543156
Paul, C., Manero, F., Gonin, S., Kretz-Remy, C., Virot, S., & Arrigo, A. P. (2002). Hsp27 as a negative regulator of cytochrome C release. Mol Cell Biol, 22(3):816-34. https://doi.org/10.1128/MCB.22.3.816-834.2002, PMid:11784858, PMCid:PMC133538
Ungelenk, S., Moayed, F., Ho, C. T., Grousl, T., Scharf, A., Mashaghi, A., et al. (2016). Small heat shock proteins sequester misfolding proteins in near-native conformation for cellular protection and efficient refolding. Nature Communications, 7, Article number: 13673. https://doi.org/10.1038/ncomms13673,
PMid:27901028, PMCid:PMC5141385
Wang, T., Maser, P., & Picard, D. (2016). Inhibition of Plasmodium falciparum Hsp90 Contributes to the Antimalarial Activities of Aminoalcohol-carbazoles. Med. Chem., 59(13), 6344–6352. https://doi.org/10.1021/acs.jmedchem.6b00591, PMid:27312008
Sittler, A., Lurz, R., Lueder, G., Priller, J., Lehrach, H., Hayer-Hartl, M. K., et al. (2001). Geldanamycin activates a heat shock response and inhibits huntingtin aggregation in a cell culture model of Huntington’s disease. Human Molecular Genetics, 10(12), 1307-1315. https://doi.org/10.1093/hmg/10.12.1307,
PMid:11406612
Jurczyszyn, A., Zebzda, A., Czepiel, J., Perucki, W., Bazan-Socha, S., Cibor, D., et al. (2014). Geldanamycin and Its Derivatives Inhibit the Growth of Myeloma Cells and Reduce the Expression of the MET Receptor. J Cancer, 5(6), 480–490. https://doi.org/10.7150/jca.8731, PMid:24959301, PMCid:PMC4066360
Li, Y., Zhang, T., & Sun, D. (2009). New developments in Hsp90 inhibitors as anti-cancer therapeutics: mechanisms, clinical perspective and more potential. Drug Resist Updat, 12(1-2), 17–27. https://doi.org/10.1016/j.drup.2008.12.002,
PMid:19179103, PMCid:PMC2692088
Karwa, R., & Wargo, K. A. (2009). Efungumab: a novel agent in the treatment of invasive candidiasis. Ann Pharmacother, 43(11), 1818-1823. https://doi.org/10.1345/aph.1M218,
PMid:19773528
Phillips, P. A., Dudeja, V., McCarroll, J.A., Borja-Cacho, D., Dawra, R. K., Grizzle, W. E., et al. (2007). Triptolide induces pancreatic cancer cell death via inhibition of heat shock protein 70. Cancer Res, 67(19), 9407-9416. https://doi.org/10.1158/0008-5472.CAN-07-1077, PMid:17909050
Jacobson, B. A., Chen, E. Z., Tang, S., Belgum, H. S., McCauley, J. A., Evenson, K. A., et al. (2015). Triptolide and its prodrug minnelide suppress Hsp70 and inhibit in vivo growth in a xenograft model of mesothelioma. Genes Cancer, 6(3-4), 144–152. PMid:26000097, PMCid:PMC4426951
O'Leary, J. C., Li, Q., Marinec, P., Blair, L. J., Congdon, E. E., Johnson, A. G., et al. (2010). Phenothiazine-mediated rescue of cognition in tau transgenic mice requires neuroprotection and reduced soluble tau burden. Molecular Neurodegeneration, 5(1), Article 45. https://doi.org/10.1186/1750-1326-5-45,
PMid:21040568, PMCid:PMC2989315
Chang, C. L., Hsu, Y. T., Wu. C. C., Yang, Y. C., Wang, C., Wu, T. C., et al. (2012). Immune mechanism of the antitumor effects generated by bortezomib. J Immunol, 189(6), 3209-3220. https://doi.org/10.4049/jimmunol.1103826,
PMid:22896634
Ekimov, I. V., & Plaksina, D. V. (2017). Effects of Quercetin on Neurodegenerative and Compensatory Processes in the Nigrostriatal System in a Model of the Preclinical Stage of Parkinson’s Disease in Rats. Neuroscience and Behavioral Physiology, 47(9), 1029–1036. https://doi.org/10.1007/s11055-017-0508-x
McConnell, J. R., & McAlpine, S. R. (2013). Heat shock proteins 27, 40, and 70 as combinational and dual therapeutic cancer targets. Bioorg Med Chem Lett, 23(7), 1923–1928. https://doi.org/10.1016/j.bmcl.2013.02.014,
PMid:23453837, PMCid:PMC3602338
Chi, K. N., Yu, E. Y., Jacobs, C., Bazov, J., Kollmannsberger, C., Higano, C.S., et al. (2016). A phase I dose-escalation study of apatorsen (OGX-427), an antisense inhibitor targeting heat shock protein 27 (Hsp27), in patients with castration-resistant prostate cancer and other advanced cancers. Ann Oncol, 27(6), 1116-1122. https://doi.org/10.1093/annonc/mdw068,
PMid:27022067
Vidyasagar, A., Wilson, N. A., & Djamali, A. (2012). Heat shock protein 27 (HSP27): biomarker of disease and therapeutic target. Fibrogenesis & Tissue Repair, 5(1), 7. https://doi.org/10.1186/1755-1536-5-7,
PMid:22564335, PMCid:PMC3464729
Lee, H. J., Ock, C.Y., Kin, S. J., & Hahm, K. B. (2010). Heat Shock Protein: Hard Worker or Bad Offender for Gastric Diseases. International Journal of Proteomics, 2010, 1-11. https://doi.org/10.1155/2010/259163,
PMid:22084675, PMCid:PMC3195352
Moudgil, K. D., Thompson, S. J., Geraci, F., Paepe, B. D., & Shoenfeld, Y. (2013). Heat-Shock Proteins in Autoimmunity. Autoimmune Diseases, 2013; 1-3. https://doi.org/10.1155/2013/621417
Xu, Q. (2002). Role of Heat Shock Proteins in Atherosclerosis. Atherosclerosis, Thrombosis and Vascular biology, 22, 1547-1559. https://doi.org/10.1161/01.ATV.0000029720.59649.50
Calderwood, S.K., & Gong, J. (2016). Heat Shock Proteins Promote Cancer: It's a Protection Racket. Trends in Biochemical Sciences, 41(4), 311-323. https://doi.org/10.1016/j.tibs.2016.01.003,
PMid:26874923, PMCid:PMC4911230
Terao, A., Steininger, T. L., Hyder, K., Apte-Deshpande, A., Ding, J., Rishipathak, D., et al. (2003). Differential increase in the expression of heat shock protein family members during sleep deprivation and during sleep. Neuroscience, 116(1), 187-200. https://doi.org/10.1016/S0306-4522(02)00695-4
Read the full article
0 notes
Text
Application of In Situ Generated Chiral Oxazaborolidine Catalyst for the Enantioselective Reduction of Prochiral Ketones
INTRODUCTION
Asymmetric reduction of prochiral ketones to give enantiopure alcohols is an essential transformation with diverse applications in pharmaceutical as well as chemical industry. Although an extensive study has already been done in this area of asymmetric conversion, there remains always a need to explore more ecofriendly, stable, cheaper and easily available catalyst systems.
Undoubtedly, biocatalysis, metal catalysis and organocatalysis are regarded as three major pillars of modern asymmetric chemistry. Organocatalysis fascinates the synthetic organic chemist due to its easy availability, less toxicity and cost effectiveness.
The oxazaborolidine catalyst systems prepared from chiral amino alcohols and borane source have been extensively studied for the asymmetric reduction1-3. The parent catalyst H-CBZ (1) in situ synthesized from α,α-diphenylpyrrolidine methanol and BH3-THF has been found effective to furnish the asymmetric transformation, however due to its relatively less stability to air and moisture its corresponding B-methyl derivative (2) is favored.
Several other amino alcohols derived from natural and unnatural sources were then synthesized and applied as catalysts for various asymmetric transformations4. Chirally pure cis-1-amino-2-indanols have also been reported for asymmetric transformation.5,6
Didier and coworkers reported (1S, 2R)-(-)-cis-1-amino-2-indanol (3) as a chiral catalyst for asymmetric reduction of acetophenone using stoichiometric amounts of 1,3,2-oxazaborolidine B-H compounds. However the authors concluded that no system was found to be efficient with catalytic amounts of ligand (studied)6.
In order to achieve high enantioselectivity the essential requirement for chiral catalyst is, it must possess substituent at one face of transition state which can block attack from that side. Considering the structural advantages of indanyl moiety during the chiral transition state Gao Yun and coworkers studied and reported cis -1-amino-2-indanol for asymmetric reduction of aromatic prochiral ketones using BH3-THF System.7
The use of borane reagents has limitations due to difficulties in their handling, transportation and storage during commercial applications despite of their commercial availability. Periasamy and coworkers reported tetraalkylammonium borohydride, methyl iodide along with (S)-α,α-diphenylpyrrolidine methanol as an efficient catalyst system for asymmetric reduction of prochiral ketones by in situ formation of oxazaborolidine catalyst at 250C in THF to their corresponding alcohols with up to 99 % ee8. In the past, Periasamy and coworkers also reported that BH3-THF can be prepared in situ using sodium borohydride and iodine in THF. However the combination of α,α-diphenylpyrrolidine methanol , sodium borohydride and iodine gave poor results in asymmetric reduction of acetophenone. The primary reason for this fact was thought to be sparing solubility of sodium borohydride in THF9. Accordingly we have undertaken a study to employ (1S, 2R)-(-)-cis-1-amino-2-indanol as a chiral catalyst along with various borohydride reagents like sodium borohydride (4) tetramethylammonium borohydride (5) tetraethylammonium borohydride (6) and tetrabutylammonium borohydride (7).
Here in we report, in situ synthesis of chiral oxazaborolidine organocatalyst by using (1S, 2R)- (-)-cis-1-amino-2-indanol, tetrabutyl-ammonium borohydride and methyl iodide to reduce prochiral ketones to the corresponding alcohols with enantiomeric excess up to 96 %.
MATERIALS AND METHOD
General Information
All reactions were carried out under inert atmosphere. Thin layer chromatography (TLC) was performed on precoated aluminium TLC plates, with detection by ninhydrin stain. Products were purified by column chromatography (Ethyl acetate and Hexane solvent system).The products were identified by spectral data (1H NMR, 13C NMR, IR and physical constants) and compared with literature values.
Enantiomeric excess was determined by HPLC using chiral column, Chiralcel-OD-H. Absolute configuration was assigned by comparison of sign of the specific rotation with that of a literature value.
The physical constants were determined using digital melting point apparatus and were observed to be uncorrected. Spectral analysis was performed by 1H-NMR spectra recorded in DMSO-d6 and CDCl3 on a Bruker Avance III, 400 MHz and Bruker Avance 300 MHz. Chemical shifts were reported as d (ppm) scale using TMS as internal standard with multiplicities and number of protons. Infrared spectra were recorded on a Perkin Elmer IR Spectrometer with nmax value reported in cm-1.
All the products obtained and discussed in this work have been reported and characterized by suitable technique such as 1H NMR, 13C NMR, IR and were compared with previously reported data.
General experimental procedure for asymmetric reduction of ketones using (1S, 2R)-(-)-cis-1-amino-2-indanol, tetrabutyl-ammonium borohydride and methyl iodide
Tetrabutylammonium borohydride (7) (5 mmol) and (1S, 2R)-(-)-cis-1-amino-2-indanol (10 mol %) in THF (5 times) were taken in a three neck RB flask. The contents were stirred at 25 -300C for about 10 min under nitrogen atmosphere. Methyl iodide (5 mmol) was added using a syringe and the reaction mixture was stirred for about 30 min. Acetophenone derivative (8a-8k) (5 mmol) in THF (5 times) was added drop wisely for about 30 min under nitrogen atmosphere. The reaction mixture was stirred till reaction completion. The mixture was carefully quenched with HCl to get pH 5.0-6.0, the organic layer was extracted with Dichoromethane (10 times). The combined organic extract was washed with brine (3 Times), dried over anhydrous Na2SO4, and the solvent was evaporated to give residue. The residue was purified on a silica gel column using hexane/ethyl acetate as eluent to furnish desired chirally pure alcohol (9a-9k).
RESULTS AND DISCUSSION
Initially various borohydride reagents like sodium borohydride (4) tetramethylammonium borohydride (5) tetraethylammonium borohydride (6) and tetrabutylammonium borohydride (7) were examined (Figure 2) by employing these reagents for the asymmetric reduction of acetophenone using (1S, 2R)-(-)-cis-1-amino-2-indanol 3 as a chiral catalyst as per the reported process, Scheme 1.

Figure: 1 Chiral Organocatalysts
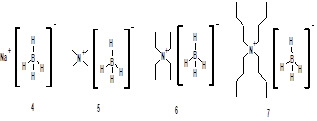
Figure: 2 Borohydride reagents examined for reduction
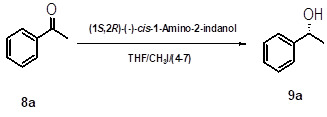
Scheme 1: Reduction of acetophenone using in situ generated oxazaborolidine catalyst
It was observed that acetophenone when subjected for asymmetric reduction by using (1S, 2R)- (-)-cis-1-amino-2-indanol (3) as a chiral catalyst with sodium borohydride and methyl iodide gave very poor enantioselectivity. Comparatively better enantioselectivity (67-73%) was obtained with tetramethylammonium borohydride (5) tetraethylammonium borohydride (6) however longer reaction time required. Employment of tetrabutylammonium borohydride (7) and methyl iodide with chiral organocatalyst (3) resulted in even better enantioselectivity (91%) with 89 % yield. The improved enantioselectivity for reagent (7) attributed to better solubility of tetrabutyl ammonium borohydride (7) in THF.
It was also observed that replacing the reaction solvent from THF to DCM or employing iodine instead of methyl iodide lowers the enantioselectivity. Initially we carried out reactions with 5 mol % of catalyst (3). When catalyst loading was increased to 10 mol %, enantiomeric excess increased upto 93 %. However further increase in catalyst loading (20 mol %) could not enhance enantioselectivity. The enantioselective reduction of acetophenone under different conditions is given in Table 1.
Table 1: Enantioselective reduction of acetophenone under different conditionsa
S. No.
Catalayst
Loading
(mol %)
Reagents
Solvent
Yield
(%)b
Enantiomeric Excessc
Absolute Configurationd
1
5
4 / CH3I
THF
85
63
(R)
2
5
5 / CH3I
THF
81
67
(R)
3
5
6 / CH3I
THF
85
73
(R)
4
5
7 / CH3I
THF
89
91
(R)
5
5
7 / Iodine
THF
87
85
(R)
6
5
7 / CH3I
DCM
86
84
(R)
7
10
7 / CH3I
THF
92
93
(R)
8
20
7 / CH3I
THF
89
93
(R)
All reactions were carried out using 5 mmol of borohydride reagent, 5 mmol of methyl iodide, 5 mmol of acetophenone, 5 - 20 mol % of (1S, 2R)-(-)-cis-1-amino-2-indanol in 5 times of solvent.
The yields are of isolated product after purification by column chromatography. The products were identified by spectral data (1H NMR, 13C NMR, IR and physical constants) and compared with literature values.
Enantiomeric excess was determined by HPLC using chiral column, Chiralcel-OD-H
Absolute configuration was assigned by comparison of sign of the specific rotation with that of a literature value.
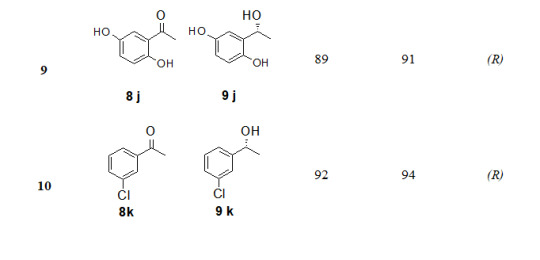
The substrate scope of this protocol is studied by varying substituent on aromatic ring of acetophenone, Table 2. Better enantioselectivity was obtained with acetophenone derivatives bearing electron withdrawing groups compared to electron donating substituents.
Table 2: Substrate scope for asymmetric reduction of ketonesa
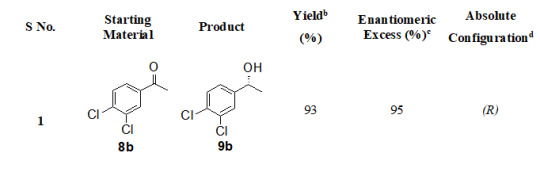
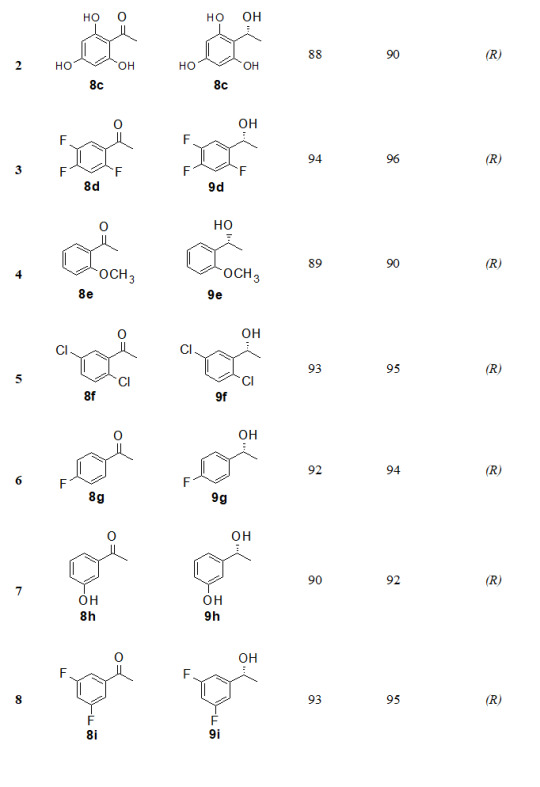
All reactions were carried out using 5 mmol of tetrabutylammonium borohydride, 5 mmol of methyl iodide, 5 mmol of acetophenone and 10 mol % of (1S,2R)-(-)-cis-1-amino-2-indanol in 5 times THF.
The yields are of isolated product after purification by column chromatography. The products were identified by spectral data (1H NMR, 13C NMR, IR, and physical constants) and compared with literature values.
Enantiomeric excess was determined by HPLC using chiral column, Chiralcel-OD-H
Absolute configuration was assigned by comparison of sign of the specific rotation with that of a literature value.
9
89
91
(R)
10
92
94
(R)
a. All reactions were carried out using 5 mmol of tetrabutylammonium borohydride, 5 mmol of methyl iodide, 5 mmol of acetophenone and 10 mol % of (1S,2R)-(-)-cis-1-amino-2-indanol in 5 times THF.
b. The yields are of isolated product after purification by column chromatography. The products were identified by spectral data (1H NMR, 13C NMR, IR, and physical constants) and compared with literature values.
c. Enantiomeric excess was determined by HPLC using chiral column, Chiralcel-OD-H
d. Absolute configuration was assigned by comparison of sign of the specific rotation with that of a literature value.
CONCLUSION
In conclusion, we have explored in situ synthesis of chiral oxazaborolidine catalyst by using (1S, 2R)-(-)-cis-1-amino-2-indanol, tetrabutylammonium borohydride and methyl iodide. Various borane reduction systems have been studied and reported in this work. The chiral organocatalyst (1S,2R)-(-)-cis-1-amino-2-indanol along with tetrabutylammonium bromide, methyl iodide was employed for the reduction of a number of ortho, meta and para substituted acetophenones. It was inferred that acetophenone derivatives bearing electron withdrawing groups depicted better enantioselectivity compared to electron donating substituents.
ACKNOWLEDGEMENT
We are thankful to the analytical department of Sun Pharmaceutical Industries Limited, Gurgaon, India for their characterization
analysis support.
REFERENCES
(a) Singh, V. K. (1992). Practical and useful methods for the enantioselective reduction of unsymmetrical ketones. Synthesis, 1992(7), 605-617. https://doi.org/10.1055/s-1992-26174
(b) Corey, E. J., & Helal, C. J. (1998). Reduction of carbonyl compounds with chiral oxazaborolidine catalysts: a new paradigm for enantioselective catalysis and a powerful new synthetic method. Angewandte Chemie International Edition, 37(15), 1986-2012. https://doi.org/10.1002/(SICI)1521-3773(19980817)37:153.0.CO;2-Z
(c) Mathre, D. J., Thompson, A. S., Douglas, A. W., Hoogsteen, K., Carroll, J. D., Corley, E. G., & Grabowski, E. J. (1993). A practical process for the preparation of tetrahydro-1-methyl-3, 3-diphenyl-1H, 3H-pyrrolo oxazaborole-borane. A highly enantioselective stoichiometric and catalytic reducing agent. The Journal of Organic Chemistry, 58(10), 2880-2888. https://doi.org/10.1021/jo00062a037
(a) Corey, E. J., Bakshi, R. K., & Shibata, S. (1987). Highly enantioselective borane reduction of ketones catalyzed by chiral oxazaborolidines. Mechanism and synthetic implications. Journal of the American Chemical Society, 109(18), 5551-5553. https://doi.org/10.1021/ja00252a056
(b) Corey, E. J., Bakshi, R. K., Shibata, S., Chen, C. P., & Singh, V. K. (1987). A stable and easily prepared catalyst for the enantioselective reduction of ketones. Applications to multistep syntheses. Journal of the American Chemical Society, 109(25), 7925-7926. https://doi.org/10.1021/ja00259a075
(a) Itsuno, S., Ito, K., Hirao, A., & Nakahama, S. (1983). Asymmetric reduction of aromatic ketones with the reagent prepared from (S)-(–)-2-amino-3-methyl-1, 1-diphenylbutan-1-ol and borane. Journal of the Chemical Society, Chemical Communications, (8), 469-470. https://doi.org/10.1039/C39830000469
(b) Corey, E. J. (1990). New enantioselective routes to biologically interesting compounds. Pure and applied chemistry, 62(7), 1209-1216. https://doi.org/10.1351/pac199062071209
Reddy, U. V. S., Chennapuram, M., Seki, C., Kwon, E., Okuyama, Y., & Nakano, H. (2016). Catalytic Efficiency of Primary β‐Amino Alcohols and Their Derivatives in Organocatalysis. European Journal of Organic Chemistry, 2016(24), 4124-4143. https://doi.org/10.1002/ejoc.201600164
Thompson, W. J., Fitzgerald, P. M.,
Holloway, M. K., Emini, E. A., Darke, P. L., McKeever, B. M., ... & Zugay, J. A. (1992). Synthesis and antiviral activity of a series of HIV-1 protease inhibitors with functionality tethered to the P1 or P1'phenyl design. Journal of medicinal chemistry, 35(10), 1685-1701. https://doi.org/10.1021/jm00088a003, PMid:1588551
Didier, E., Loubinoux, B., Tombo, G. H. R., & Rihs, G. (1991). Chemo-enzymatic synthesis of 1, 2-and 1, 3-amino-alcohols and their use in the enantioselective reduction of acetophenone and anti-acetophenone oxime methyl ether with borane. Tetrahedron, 47(27), 4941-4958. https://doi.org/10.1016/S0040-4020(01)80959-5
Hong, Y., Gao, Y., Nie, X., & Zepp, C. M. (1994). cis-1-amino-2-indanol in asymmetric synthesis. Part I. A practical catalyst system for the enantioselective borane reduction of aromatic ketones. Tetrahedron letters, 35(36), 6631-6634. https://doi.org/10.1016/S0040-4039(00)73453-8
Anwar, S., & Periasamy, M. (2006). A convenient method for the preparation of oxazaborolidine catalyst in situ using (S)-α, α-diphenylpyrrolidinemethanol, tetrabutylammonium borohydride, and methyl iodide for the asymmetric reduction of prochiral ketones. Tetrahedron: Asymmetry, 17(23), 3244-3247. https://doi.org/10.1016/j.tetasy.2006.11.032
Periasamy, M., Kanth, J. B., & Prasad, A. B. (1994). Convenient procedures for the asymmetric reductions utilizing α, α-diphenyl-pyrrolidinemethanol and borane complexes generated using the I2/NaBH4 system. Tetrahedron, 50(21), 6411-6416. https://doi.org/10.1016/S0040-4020(01)80657-8
Read the full article
0 notes
Text
In-Vitro Anthelmintic Activity Of Ehretia laevis bark On Indian Adult Earthworm
INTRODUCTION
Parasitic infection including Helminthiasis is a critical serious problem in the tropical regions including the Asian and African countries which affects more than 2.5 billions of people worldwide. Helminths produce serious problem in human beings and other animals around the world specifically to the third world countries1. Different type of helminths infects the human and animals out of which intestinal round worms Pheritima posthuma (Annelida) are most common. Approximately 200 million people suffer severe morbidity associated with these parasites and half of which are school-going children affected by massive infections. Variety of several clinical symptoms arises due to this infection include dysentery, diarrhoea, nausea, vomiting, loss of appetite, loss of weight, acidity and anaemia. Other sign and symptoms of helminthic infections include respiratory symptoms, dermatological consequences and epilepsy as a result of neurocysticercosis. Helminthic infections may also subvert immune responses to pathogens of other diseases such as tuberculosis, HIV, and malaria2. Although the majority of infections are due to the worms generally limited to tropical regions, they can also occur to travellers who have visited those areas and some of them can develop in temperate climates3. Helminthiasis is a disease in which a part of the body is infested with worms like as pinworm, roundworm or tapeworm. Typically, the worms present in the gastrointestinal tract but may also reside into the liver and other organs, infected peoples are excrete helminth eggs in their faeces, which then contaminate the soil in areas with inadequate sanitation4. Other peoples can be infected by ingesting eggs or larvae in contaminated food, or through penetration of the skin by infective larvae in the soil (hookworms). Parasitic diseases can cause severe morbidity, including filariasis (a cause of elephantiasis), onchocerciasis (river blindness), and schistosomiasis5. As per the WHO survey only synthetic drugs are sometimes used in the treatment of helminth infestations in human beings but these synthetic drugs are out of reach of millions of people and have a lot of side effect. In view of this, an attempt has been made to study the anthelmintic activity of herbal drug. Development of resistance to most of the commercially available anthelmintics drugs are became a severe problem worldwide. Sometimes, these drugs are unaffordable, inaccessible or inadequately available to the resource poor farmers of the developed and developing countries6. These factors paved the way for herbal remedies as alternative anthelmintics7. Therefore the evaluation of the activities of medicinal plants claimed for possessing the anthelmintic property is getting attention these days8. Screening and proper evaluation of the claimed medicinal plants as anthelmentics could offer possible alternatives that may be both sustainable and environmentally acceptable9. Ehretia laevis is fast-growing small tree belonging to family Ehretiaceae. The plant is native to India, Pakistan, Laos, Myanmar, Vietnam, China, Bhutan. The plant Ehretia laevis is located at hilly forests, in ravine and on hill slopes. The plant is known as Dant-Rang, Vadhvarni, Chamror10. The inner bark of E. laevis is used as food. Leaves are applied to ulcers, skin diseases and in headache. Fruit is used as urinary passage, lung and spleen diseases, astringent, anthelmintic, diuretic, demulcent, expectorant. Powdered kernel mixed with oil is a remedy in ringworm. Seeds are anthelmintic. Barks are used in throat infection. Root for veneral diseases. The plant contains chemical constituents likes fatty acids, phenolic acids, flavonoids, cyanogenetic glycosides, and benzoquinones11,12.
In the current study, we have attempted to investigate methanolic, hydroalcoholic and aqueous extracts of bark of medicinal plant Ehretia laevis for their claimed anthelmintic activity.
MATERIALS AND METHOD
Plant Collection
The fresh barks of plant Ehretia laevis were collected from haripura and manudevi region of Taluka Yawal, District Jalgaon, India. The selected plants were authenticated by Dr. D. A. Dhale, Asst. Professor, PG & Research Dept. of Botany SSVPS’s, L.K.Dr.P.R.Ghogrey Science College, Dhule, Maharashtra. Barks were dried at room temperature to avoid loss of chemical constituents and milled with the aid of grinding machine.
Selection of Experimental Worms
Indian adult earthworms (Pheretima posthuma) were used to carry out the experiment. Pheritima posthuma is commonly known as earthworm and were collected from water logged areas. Ascardia galli is nematode were obtained from freshly slaughtered area. Both worms were identified by PG Department of Zoology, SSVPS's Science College, Dhule. Worms were washed with normal saline to remove all faecal matter. The earthworms of 7-9 cm in length and 0.2-0.4 cm in width were used for all the experimental protocol. Ready availability, anatomical and physiological resemblance of Pheretima posthuma and Ascardia galli made it to be used initially for in-vitro evaluation of anthelmintic activity.
Preparation of Plant extract
The bark of plant were thoroughly washed with tap water, dried at room temperature and transformed to coarse powder. The bark powder were extracted with three solvents i.e methanol, water and water-ethanol separately by Soxhlet extraction method. Finally, the extract was evaporated and dried under vacuum to obtain thick sticky extract.
Drugs and Chemicals
Piperazine citrate , Methanol, Distilled water, Ethanol and were used during the expperimental protocol. All the chemicals used are laboratory and analytical grade.
Experimental Work13,14,15,16
The anthelmintic activity was carried out as described by Ajaiyeoba EO. et. al, 2001, with minor modifications. The assay was performed on adult Indian earthworm Pheritima posthuma and Ascardia galli due to their anatomical and physiological resemblance with the intestinal round worm parasite of human being17,18. Because of easy availability, earth worms have been used widely for initial evaluation of anthelmentic compounds in vitro. The Indian earthworm Pheritima posthuma and Ascardia galli, of nearly equal size, six in each group was taken for the experiment. The methanolic, aqueous and hydroalcoholic dried extract were suspended in 1% w/v Carboxy Methyl Cellulose, prepared in normal saline water in three different conc. (10, 25 and 50 mg/ml). Piperazine citrate suspension of concentration 10mg/ml was taken as standard and normal saline water with 1% CMC was taken as a control. Worms were placed in petridish containing 25 ml of sample (drug) solution. Time for paralysis was noted either when any movement could not be observed except when the worms were shaken vigorously or when dipped in warm water (500C). Death was included when the worms lost their motility followed by white secretions and fading away of their body colour.
Statistical Analysis19
The data presented as Mean ± SEM. The activities of both the leaves extracts were compared with the control. All the extracts showed significantly higher duration of paralysis and death. Values of P
Read the full article
0 notes
Text
Novel Poly(Vinyl caprolactum-co-Sodiumacrylate) Microspheres for Controlled Release of 5-Fluorouracil
INTRODUCTION
Controlled delivery of drugs by means of biodegradable polymers began in the 1970s and continued to expand rapidly with numerous novel products1,2. The controlled release technology has lead to the development of newer methods of drug administration as well as the design and application of different types of CR formulations for effective targeting of certain drugs to the site of action. In particular, biodegradable polymeric systems have led to the development of CR dosage formulations to achieve the desired therapeutic results to obtain maximum dose regimen with minimum side effects3. The release of drug from a polymer matrix occurs due to the transport of drug to the surrounding medium system by the molecular diffusion mechanism. The CR systems offer many advantages over the conventional dosage forms, including improved efficacy, reduced toxicity as well as improved patient compliance and convenience4-6. Among the various types of polymers employed, hydrophilic biopolymers are quite suitable for oral applications7 due to their several inherent advantages over the synthetic polymers.
Drug targeting to a specific tissue or organ has been the subject of creative and innovative research in medicinal and pharmaceutical chemistry since the beginning of the twentieth century. In many diseases (e.g. cancer, AIDS, rheumatoid arthritis, etc.) a considerable therapeutic advantage could be gained if drugs were delivered more selectively and in a controlled manner to their target sites. More particularly, it is conventionally accepted that efficient, compliant and reliable therapy requires that the drug reside as long as its therapeutic action is needed at a specific site, where it acts (by systemic absorption, binding, inhibition, etc.) as intact molecules. This concept has led to the development of a variety of physically based controlled release dosage forms such as drug dispersible matrices, coated tablets or particles, microcapsules. The development of an appropriate delivery system will first require a proper consideration of three related factors; the properties of the drug; the disease and the destination in the body.
Over the past few years, stimuli-responsive (sensitive) polymers have become the object of intensive study due to their ability to change drastically their physical state under minute changes in external environment such as temperature, pH, ionic strength, light illumination, etc. Recently, chromatographic8,9 drug delivery10,11 membrane technology12,13 and kinetic inhibition14 applications were reported. Poly(N-isopropylacrylamide)15-17 (PNIPA) and poly(N-vinyl caprolactam)18,19 (PVCL) were intensively investigated due to their thermo-sensitive properties since these are water soluble at low temperature. However, they exhibit lower critical solution temperature (LCST) in water and undergo a coil-to-globule transition and aggregation at higher temperatures. For PNIPA the coil-to-globule transition occurs at around 32°C. PVCL is a homolog of poly(N-vinylpyrrolidone) (PVP), which is a biocompatible polymer widely used in medicine and pharmaceutics20. PVCL combines the useful and important properties of PVP and PNIPAm. It is a biocompatible polymer with a phase transition in the region of physiological temperature (30-37 °C). Such properties make it a prospective material in designing CR systems. Further, the incorporation of ionic hydrophilic moieties into the PolyVCL hydrogel networks would enhance the LCST and the gels become sensitive towards PH, whereas hydrophobic moieties decrease the LCST. Liu et al.21 found that salts of acrylic acid monomers are strong electrolytes, which are completely ionized in water, and their copolymeric units increased the swelling characteristics to a greater extent.
5-Fluorouracil is an acidic, water-soluble22,23, hydrophilic, is an antineoplastic drug used extensively in clinical chemotherapy for the treatment of solid tumors. It has been widely used in drug administration due to its large number of secondary effects that accompany its conventional administration. We present here the development of 5-fluorouracil-loaded poly(vinyl caprolactam-co-Sodium acrylate) microspheres for investigating its slow release characteristics. The plasma lifetime of 5-Fu is 1-1.2 hand it needs to extend for its effective therapy. The microspheres prepared were characterized by particle size analyzer, differential scanning calorimetry (DSC) and scanning electron microscopy (SEM). The in vitro release studies have been performed in 7.4 pH buffer solution at 25 0C and 370C to extend to the release rates of the drug.
MATERIALS AND METHOD
Materials
Vinyl caprolactam (VC) was purchased from Aldrich Chemicals, Milwaukee, WI USA. Sodium acrylate (SA), N, N¢-methylene bisacrylamide (NNMBA), sodium lauryl sulfate, potassium persulfate, and calcium chloride were all purchased from s.d. fine chemicals, Mumbai, India. 5-Fluorouracil was purchased from MP Biochemicals, Eschwege, Germany.
Synthesis of poly(vinyl caprolactam-co-sodium acrylate) microspheres
Sodium lauryl sulfate (1g) was dissolved in 80 ml of water taken in a three-necked round bottom flask equipped with a mechanical stirrer, a condenser, and a gas inlet to maintain the inert nitrogen atmosphere. The flask was immersed in an oil bath with a thermostatic control to maintain the desired temperature accurate to ± 1oC. The solution was stirred at 800 rpm speed until it became clear and 100 mg of potassium persulfate was added. The required amount of SA, VC, crosslinking agent, NNMBA and 5-Fluorouracil were dissolved separately in 20 ml of water. This mixture was added to the reaction mixture drop-wise using a dropping funnel and the reaction was continued for 8 h at 700C to obtain the maximum yield. The reaction mixture was taken out after 8 h and added to 1% calcium chloride solution drop-wise to break the emulsion24. Particles were then isolated by centrifuging the product at the rotor speed of 12,000 rpm, washed with water and dried under vacuum at 400C for 24 h.
Conversion of Copolymer
The yield of copolymeric microspheres was determined gravimetrically. After copolymerization, the latex solution was added to 1 % calcium chloride solution and centrifuged to isolate the particles from the mixture. The copolymeric microspheres were washed several times successively with water and methanol solvents to remove the remaining monomer and initiator and then dried in a vacuum oven at 500C until attainment of constant weight. The % conversion of monomers was calculated as:
% Conversion = (W/M) ×100
Where W is the weight of the dry copolymer obtained from the latex sample and M is the weight of the monomers taken. The yield of copolymeric microspheres varied between 80 and 85 % for various formulations prepared in this study.
pH and Temperature Sensitive Nature of Copolymer Microspheres

percentages of swelling ratio (% SR)
pH and temperature sensitivity of copolymer microspheres were studied through swelling experiments. First, the microspheres were immersed in a buffer solution with various pH values (pH buffer solutions were prepared using NaH2PO4, Na2HPO4, NaCl and NaOH solution and pH values were measured using ELICO pH meter, India) at 30oC for 12 h. The swollen MGs were taken out for every 30 min and removed surface adhered buffer solution using tissue paper. The MGs were further immersed in various buffer solutions to reach equilibrium swelling. Swelling experiments were carried out in water by mass measurements at various temperatures to study temperature responsive behavior of microspheres. The percentages of swelling ratio (% SR) were calculated using the following equations.
Where, Ws is the weight of swollen gel at time t, and Wd is the dry weight of the hydrogel. Mass measurements were made on a digital ADAMS microbalance (Model AF 210L, U.K) with a sensitivity of 0.01 mg. Each value was averaged over three parallel measurements. Statistical analysis was performed using one-way ANOVA way in ORIGIN 8.0. All quantitative data are presented as means + standard deviation.
Differential Scanning Calorimetry (DSC) Studies
Differential scanning calorimetric (DSC) curves were recorded on a Rheometric scientific differential scanning calorimeter (Model-DSC SP, UK). The instrument was calibrated using indium as the standard. Samples were heated in sealed aluminum pans between 300 and 400oC at the heating rate of 10oC/min under inert nitrogen purge gas at the rate of 20 ml/min.
Scanning Electron Microscopic (SEM) Studies
Morphology of the microspheres was confirmed by scanning electron microscopy (SEM). Micrographs of the dry microspheres in powder form, dispersed in acetone, were all recorded using Leica 400, Cambridge, UK instrument. Particle Size Analysis
Size distribution of the microspheres was determined using the particle size analyzer (Mastersizer 2000, Malvern Instruments, UK) equipped with the dry accessory system.
Estimation of Drug Loading and Encapsulation Efficiency
Loading efficiency of 5-FU in the microspheres was determined spectrophotometrically. About 10 mg of the drug-loaded core-shell microspheres were placed in 10 ml of buffer solution and stirred vigorously for 48 h to extract the drug from the microspheres. The solution was filtered and assayed by UV spectrophotometer (model Anthelme, Secomam, Dumont, France) at the fixed lmax value of 270 nm. The results of % drug loading and encapsulation efficiency were calculated, respectively using Equations. (1) and (2). These data are compiled in Tables 1 and 2, respectively.
Table 1: Results of % encapsulation efficiency and mean diameter of poly(VC-co-SA) microspheres with different amounts of crosslinking agent, monomer concentration and 5-fluorouracil
Sample code
% Vinyl Caprolactum
(VC)
% SA
% NNMBA
% 5-FU
% Encapsulation efficiency ± SD
Mean particle diameter (mm) ± SD
VCSA-1
20
80
1
5
70 ± 1
29 ± 6
VCSA-2
20
80
1
10
74 ± 2
31 ± 8
VCSA-3
20
80
1
15
78 ± 2
34 ± 6
VCSA-4
20
80
2
10
75 ± 9
28 ± 4
VCSA-5
20
80
3
10
71 ± 8
16 ± 2
VCSA-6
10
90
1
10
68 ± 6
30 ± 4
VCSA-7
30
70
1
10
71 ± 5
24 ± 1
VCSA-8
00
100
1
10
72 ± 1
22 ± 8
Table 2: Release kinetics parameters of microspheres with different amounts of crosslinking agent, monomer concentration and 5-fluorouracil at 370C
Formulation codes
K x 102
n
Correlation coefficient ‘r’
VCSA-1
0.008
0.74
0.972
VCSA-2
0.023
0.57
0.999
VCSA-3
0.026
0.55
0.999
VCSA-4
0.021
0.57
0.996
VCSA-5
0.011
0.66
0.971
VCSA-6
0.014
0.64
0.979
VCSA-7
0.011
0.71
0.978
VCSA-8
0.027
0.59
0.990
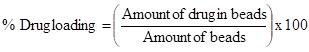
% Drug Loading
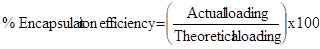
$ Encapsulation Efficiency
In-vitro Release Study
Dissolution was carried out using Tablet dissolution tester (Lab India, Mumbai, India) equipped with eight baskets. Dissolution rates were measured at 370C under 100 rpm speed. Drug release from the microspheres was studied in 7.4 pH phosphate buffer solution. Aliquot samples were withdrawn at regular time intervals and analyzed by UV spectrophotometer as explained before.
RESULTS AND DISCUSSION
pH and Temperature Responsive Behavior of Microspheres
Figure 1 (a) shows the swelling ratio of microspheres at various pH solutions. As we can clearly see that the swelling ratio of microspheres slowly increases when pH increases up to 5.0 after that it increases rapidly up to pH 8. Because at low pH i.e.,
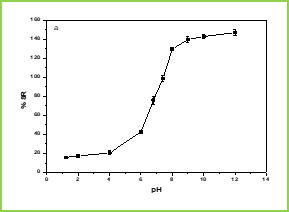
Figure 1.(a)

Figure 1.(b)
Figure 1. Swelling studies of MGs (a) various pH conditions, and (b) different temperatures
The effect of temperature on the equilibrium swelling ratios for microspheres is shown in Figure 1(b) The swelling ratio of microspheres is higher at low temperature ( LCST). This is because below LCST VCL contains a hydrophilic group (-CONH-) and hydrophobic isopropyl group present in the linear polymer chain. So, the hydrophilic group in the polymer structure will form an intermolecular hydrogen bond with surrounding water at low temperature (below the gel transition temperature); above LCST the hydrogen bonds are broken and the water molecules are expelled from the polymer. These two results make the water molecule inside the gel change from a bound state to a free State and release from the gel. This phenomenon makes the swelling ratios of the microspheres decrease rapidly at the gel transition temperature.
Differential scanning calorimetry (DSC)
DSC tracings of pure 5-fluorouracil, drug-loaded microspheres, and plain microspheres are displayed in Figure 2. The pure 5-FU exhibits a sharp peak at 285oC (curve c) is due to polymorphism and melting. However, this peak has not appeared in the case of drug-loaded microspheres (curve b) which confirms that the drug is molecularly dispersed in the polymeric microspheres.

Figure 2: DSC thermograms of (a) plain Poly(VC-co-SA) microspheres (c) 5-FU loaded Poly(VC-co-SA) microspheres and (c) 5-FU

Figure 3: Scanning electron micrographs of Poly(VC-co-SA) microspheres
Scanning Electron Microscopic (SEM) Studies
Figure 3. shows the morphology of microspheres. The formed copolymer particles are spherical with the diameters of around 10 mm.
Laser Particle Size Analyzer
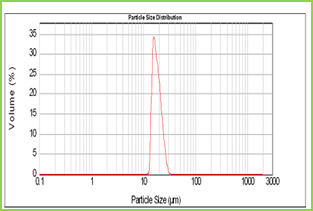
Figure 4: Particle size distribution curve of Poly(VC-co-SA) microspheres
Results of the mean particle size with standard errors are presented in Table 1, while the size distribution curve for a typical formulation containing SA-5 is displayed in Figure 4.
It is found that size distribution is broad and volume means diameter of the particle is around 16 mm. The particle size of different formulations containing different amounts of drug, crosslinking agent and different ratios of VC-co-SA are given in Table 1. The particle size of formulations containing different amounts of crosslinking agent (NNMBA) i.e., 1, 2 and 3 % are 34, 28 and 16, respectively. The particle size decreased with increasing amount of crosslinking due to the formation of a rigid structure due to a reduction in chain length of the polymer formed.
Encapsulation Efficiency
Results of encapsulation efficiencies are given in Table 1. The % encapsulation efficiency varied depending upon the initial loading of the drug. In general, for formulations VCSA-1, VCSA-2 and VCSA-3, the % encapsulation efficiency increased systematically with increasing drug content of the matrices. At higher amount of crosslinking agent i.e., 2 % or 3 % of NNMBA in the matrix, the % encapsulation efficiency decreased. The highest % encapsulation efficiency of 79 was observed for VCSA-3 containing 15 % of 5-FU with a higher amount of SA in the copolymer matrix and its size was also highest i.e., 34 mm.
Drug Release Kinetics
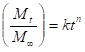
cumulative release data
While studying the drug release from the polymer matrices, it has been the usual practice to analyze the release data using the empirical relationship proposed by Ritger and Peppas25. In the present study, we have analyzed the cumulative release data using26.
Here, the ratio, Mt/M∞ represents the fractional drug release at the time, t; k is a constant characteristic of the drug-polymer system and n is an empirical parameter characterizing the release mechanism. Using the least-squares procedure, we have estimated the values of n and k for all the nine formulations at a 95% confidence limit; these data are given in Table 2 at 370C. If the values of n = 0.5, then drug diffuses and releases out of the microsphere matrix following the Fickian diffusion. If n > 0.5, anomalous or non-Fickian transport occurs. For n = 1, non-Fickian or more commonly called Case II release kinetics is operative. The values of n ranging between 0.5 and 1 indicate the anomalous type transport27.
The values of k and n have shown a dependence on the extent of crosslinking, % drug loading and SA content of the matrix. Values of n for microspheres prepared by varying the amount of SA 90, 80 and 70 % in the microspheres of by keeping 5-FU (10 %) and 1 % NNMBA, ranged from 0.70 to 0.56 leading to a shift of transport from Fickian to anomalous type. The 5-FU-loaded particles have the n values ranging from 0.55 to 0.73, indicating the shift from erosion type release to a swelling-controlled non-Fickian type of mechanism. This could be possibly due to a reduction in the regions of low microviscosity and closure of microcavities in the swollen state. Similar findings have been observed elsewhere, wherein the effect of different polymer ratios on dissolution kinetics was observed. On the other hand, the values of k are quite smaller for drug-loaded microspheres, suggesting their lesser interactions compared to microspheres containing varying amount of SA.
Effect of Sodium Acrylate Content
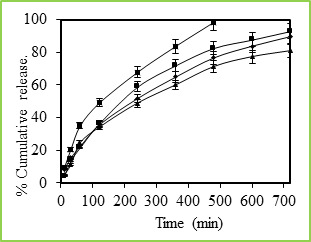
Figure 5: % Cumulative release of 5-fluorouracil through Poly(VC-co-SA) microspheres containing different amount of acrylamide at 37 0C, Symbols: (■)100 %, (■)30 %, (•) 20 % and (▲) 10 %
Figure 5 shows the in vitro release data of 5-fluorouracil from poly(VC-co-SA) particles performed with particles taking the different ratio of SA. These data show that higher amount of SA containing particles have more encapsulation efficiencies and also release studies have shown that higher amounts SA containing particles have shown prolonged release characteristics than the microspheres containing lower amounts of SA. Generally, the drug release pattern depends upon factors like particle size, crystallinity, surface character, molecular weight, polymer composition, swelling ratio, degradation rate, drug binding affinity, the rate of hydration of polymeric materials, etc.27. In the release behavior of poly(VC-co-SA) system, one can consider the binding affinity of drug and polymer swelling property of SA. A rapid release of more than 98% of the drug was observed within 12 h. from the microspheres containing a lower amount of SA, indicating on the interaction between the two polymers.
Effect of Temperature
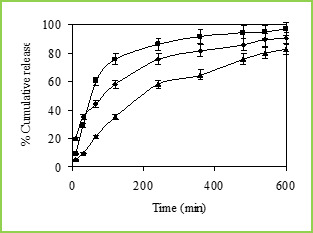
Figure 6: % Cumulative release of 5-fluorouracil through Poly(VC-co-SA) microspheres containing different amount of Vinyl Caprolactum at 25 0C, Symbols: (■)10 %, (•) 20 % and (▲) 30 %.
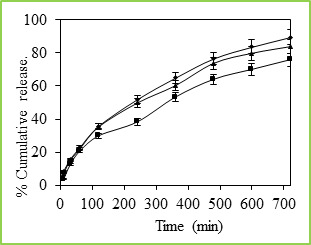
Figure 7: % Cumulative release of 5-fluorouracil through Poly(VC-co-SA) microspheres containing different amount of crosslinking agent at 37 0C, Symbols: (■) 3%, (▲) 2% and (•) 1 %
The cumulative release data vs time curves for varying amounts of vinyl caprolactam are displayed in Figure 6 at 250C. Drug release profiles exhibited drastic changes by variations in temperature from 370 to 250C as shown in Figures 4 and 5, respectively. It may be noticed that drug was released slowly at 370C i.e., above the LCST of 320C, but the release was much faster at 250C i.e., below the LCST than at 370C. This is due to the fact that at a higher temperature, the surface of microspheres would shrink, causing the drug to migrate toward the surface of the microspheres as seen by the initial burst effect during the dissolution experiments (Figure 6 and 7). However, dense surfaces of the microspheres will prohibit the release of more amount of drug. At lower temperatures, the already shrunken surface layer starts to re-swell, which would allow the drug to be released after a certain period of time, depending upon the minimum time required for re-swelling of the surface. Thus, the time required for drug release was accelerated as a result of cooling below the LCST, which further slowed down upon reheating. Microspheres were thus found to be sensitive to changes in temperature. At 250C (in the swollen state), the release rate and the total amount of drug release were considerably higher than those found at 370C (in a collapsed state). Drug molecules entrapped inside the polymer network will diffuse out of the microspheres, since they quickly get hydrated in the swollen state. In contrast, at 370C, the network structure is collapsed and exhibits a lesser tendency to uptake water or buffer solution, leading to a decrease in drug diffusion rate.
Effect of Crosslinking Agent
The % cumulative release vs time curves for varying amounts of NNMBA are displayed in Figure 7. The % cumulative release is quite fast and large at the lower amount of NNMBA, whereas release is quite slower at a higher amount of NNMBA. The cumulative release is somewhat smaller when a lower amount of NNMBA was used probably because, at higher concentration of NNMBA, polymeric chains would become rigid due to the contraction of microvoids, thus decreasing the % cumulative release of 5-FU through the polymeric matrices. As expected, the release becomes slower at a higher amount of NNMBA but becomes faster at a lower amount of NNMBA.
Effect of Drug Concentration

Figure 8 % Cumulative release of 5-fluorouracil through Poly(VC-co-SA) microspheres containing different amount of 5-FU at 37 0C, Symbols: (■) 15 %, (•) 10 % and (▲) 5 %
Figure 8 displays the release profiles of poly(VC-co-SA) microspheres that are loaded with different amounts of 5-FU. Notice that initially, during the first hour, the release is quite fast in all the formulations, but later it slowed down. The similar findings were observed in earlier literature of 5-fluorouracil loaded microspheres of a different kind . Release data suggest that those formulations containing the highest amount of drug (i.e., 15 wt. %) displayed higher release rates than those containing smaller amounts of 5-FU (i.e., 10 and 5 wt. %).
A prolonged and slow release was observed for the formulation containing a lower amount of 5-FU (i.e., 5 wt. %) at 370C; this is due to the large free volume spaces available in the matrix through which, a lesser number of 5-FU molecules would transport. Notice that for all the 5-FU-loaded formulations, the almost complete release of 5-FU was achieved after 720 min.
CONCLUSION
Poly(vinyl caprolactam-Sodium acrylate) copolymeric microspheres crosslinked with N, N¢-methylene bisacrylamide were prepared by free radical emulsion polymerization. The microspheres have been characterized by differential scanning calorimetry (DSC) and x-ray diffractometry (x-RD) to understand the drug dispersion in microspheres. Microspheres with different copolymer compositions were prepared in yields of 80-85 %. DSC indicated a uniform distribution of 5-fluorouracil particles in microspheres, whereas SEM suggested a spherical structure of the microspheres with the slight rough surface. The in vitro drug release indicated that particle size and release kinetics depend upon copolymer composition, amount of crosslinking agent and amount of 5-fluorouracil present in the microspheres.
REFERENCES
Dunn, R. L., & Ottenbrite, R. M. (Eds.). (1991). Polymeric drugs and drug delivery systems. American Chemical Society. https://doi.org/10.1021/bk-1991-0469
El-Nokaly, M. A., Piatt, D. M., & Charpentier, B. A. (1993). Polymeric delivery systems. American Chemical Society. https://doi.org/10.1021/bk-1993-0520
Garcı́a, O., Blanco, M. D., Martı́n, J. A., & Teijón, J. M. (2000). 5-Fluorouracil trapping in poly (2-hydroxyethyl methacrylate-co-acrylamide) hydrogels: in vitro drug delivery studies. European polymer journal, 36(1), 111-122. https://doi.org/10.1016/S0014-3057(99)00037-3
Uhrich, K. E., Cannizzaro, S. M., Langer, R. S., & Shakesheff, K. M. (1999). Polymeric systems for controlled drug release. Chemical reviews, 99(11), 3181-3198. https://doi.org/10.1021/cr940351u
, PMid:11749514
Işiklan, N. (2006). Controlled release of insecticide carbaryl from sodium alginate, sodium alginate/gelatin, and sodium alginate/sodium carboxymethyl cellulose blend beads crosslinked with glutaraldehyde. Journal of applied polymer science, 99(4), 1310-1319. https://doi.org/10.1002/app.22012
Vaithiyalingam, S., Nutan, M., Reddy, I., & Khan, M. (2002). Preparation and characterization of a customized cellulose acetate butyrate dispersion for controlled drug delivery. Journal of pharmaceutical sciences, 91(6), 1512-1522. https://doi.org/10.1002/jps.10155 ,
PMid:12115850
Xing, L., Dawei, C., Liping, X., & Rongqing, Z. (2003). Oral colon-specific drug delivery for bee venom peptide: development of a coated calcium alginate gel beads-entrapped liposome. Journal of Controlled Release, 93(3), 293-300. https://doi.org/10.1016/j.jconrel.2003.08.019 ,
PMid:14644579
Hosoya, K., Kubo, T., Takahashi, K., Ikegami, T., & Tanaka, N. (2002). Novel surface modification of polymer-based separation media controlling separation selectivity, retentivity and generation of electroosmotic flow. Journal of Chromatography A, 979(1-2), 3-10. https://doi.org/10.1016/S0021-9673(02)01255-4
Kanazawa, H., Yamamoto, K., Matsushima, Y., Takai, N., Kikuchi, A., Sakurai, Y., & Okano, T. (1996). Temperature-responsive chromatography using poly (N-isopropylacrylamide)-modified silica. Analytical Chemistry, 68(1), 100-105. https://doi.org/10.1021/ac950359j ,
PMid:21619225
Vihola, H., Laukkanen, A., Hirvonen, J., & Tenhu, H. (2002). Binding and release of drugs into and from thermosensitive poly (N-vinyl caprolactam) nanoparticles. European journal of pharmaceutical sciences, 16(1-2), 69-74. https://doi.org/10.1016/S0928-0987(02)00076-3
Torres-Lugo, M., & Peppas, N. A. (1999). Molecular design and in vitro studies of novel pH-sensitive hydrogels for the oral delivery of calcitonin. Macromolecules, 32(20), 6646-6651. https://doi.org/10.1021/ma990541c
Kirsh, Y. E., Vorobiev, A. V., Yanul, N. A., Fedotov, Y. A., & Timashev, S. F. (2001). Facilitated acetylene transfer through membranes composed of sulfonate-containing aromatic polyamides and poly-N-vinylamides involving nanocluster silver. Separation and purification technology, 22, 559-565. https://doi.org/10.1016/S1383-5866(00)00138-6
Hester, J. F., Olugebefola, S. C., & Mayes, A. M. (2002). Preparation of pH-responsive polymer membranes by self-organization. Journal of Membrane Science, 208(1-2), 375-388. https://doi.org/10.1016/S0376-7388(02)00317-4
Lederhos, J. P., Long, J. P., Sum, A., Christiansen, R. L., & Sloan Jr, E. D. (1996). Effective kinetic inhibitors for natural gas hydrates. Chemical Engineering Science, 51(8), 1221-1229. https://doi.org/10.1016/0009-2509(95)00370-3
Coughlan, D. C., Quilty, F. P., & Corrigan, O. I. (2004). Effect of drug physicochemical properties on swelling/deswelling kinetics and pulsatile drug release from thermoresponsive poly (N-isopropylacrylamide) hydrogels. Journal of Controlled Release, 98(1), 97-114. https://doi.org/10.1016/j.jconrel.2004.04.014 ,
PMid:15245893
Schild, H. G. (1992). Poly (N-isopropylacrylamide): experiment, theory and application. Progress in polymer science, 17(2), 163-249. https://doi.org/10.1016/0079-6700(92)90023-R
Kirsh, Y. E., Yanul, N. A., & Kalninsh, K. K. (1999). Structural transformations and water associate interactions in poly-N-vinylcaprolactam–water system. European polymer journal, 35(2), 305-316. https://doi.org/10.1016/S0014-3057(98)00114-1
Meeussen, F., Nies, E., Berghmans, H., Verbrugghe, S., Goethals, E., & Du Prez, F. (2000). Phase behaviour of poly (N-vinyl caprolactam) in water. Polymer, 41(24), 8597-8602. https://doi.org/10.1016/S0032-3861(00)00255-X
Lozinsky, V. I., Simenel, I. A., Kurskaya, E. A., Kulakova, V. K., Galaev, I. Y., Mattiasson, B., ... & Khokhlov, A. R. (2000). Synthesis of N-vinylcaprolactam polymers in water-containing media. Polymer, 41(17), 6507-6518. https://doi.org/10.1016/S0032-3861(99)00844-7
S. Barabas In Encyclopedia of Polymer Science and Engineering, 2nd ed.; Mark, H. F., Bicales, N. M., Overberger, C. C., Menges, G., Eds.; John Wiley & Sons: New York, 1985; Vol. 17, pp 225-226.
Liu, J.L. Velada, M.B.Huglin, Polymer 40 (1999) 4299. https://doi.org/10.1016/S0032-3861(98)00458-3, https://doi.org/10.1016/S0032-3861(99)00081-6,
https://doi.org/10.1016/S0032-3861(98)00387-5,
https://doi.org/10.1016/S0032-3861(98)00533-3,
https://doi.org/10.1016/S0032-3861(99)00101-9,
https://doi.org/10.1016/S0032-3861(98)00758-7,
https://doi.org/10.1016/S0032-3861(98)00660-0,
https://doi.org/10.1016/S0032-3861(98)00858-1
Yuksel, D. E. A. (1999). Preparation of spray-dried microspheres of indomethacin and examination of the effects of coating on dissolution rates. Journal of microencapsulation, 16(3), 315-324. https://doi.org/10.1080/026520499289040,
PMid:10340217
Sairam, M., Babu, V. R., Naidu, B. V. K., & Aminabhavi, T. M. (2006). Encapsulation efficiency and controlled release characteristics of crosslinked polyacrylamide particles. International journal of pharmaceutics, 320(1-2), 131-136. https://doi.org/10.1016/j.ijpharm.2006.05.001,
PMid:16766148
Babu, V. R., Sairam, M., Hosamani, K. M., & Aminabhavi, T. M. (2006). Development of 5-fluorouracil loaded poly (acrylamide-co-methylmethacrylate) novel core-shell microspheres: In vitro release studies. International journal of pharmaceutics, 325(1-2), 55-62. https://doi.org/10.1016/j.ijpharm.2006.06.020,
PMid:16884868
Ritger, P. L., & Peppas, N. A. (1987). A simple equation for description of solute release II. Fickian and anomalous release from swellable devices. Journal of controlled release, 5(1), 37-42. https://doi.org/10.1016/0168-3659(87)90035-6
Harogoppad, S. B., & Aminabhavi, T. M. (1991). Diffusion and sorption of organic liquids through polymer membranes. 5. Neoprene, styrene-butadiene-rubber, ethylene-propylene-diene terpolymer, and natural rubber versus hydrocarbons (C8-C16). Macromolecules, 24(9), 2598-2605. https://doi.org/10.1021/ma00009a070
Ratner, B. D., Hoffman, A. S., Schoen, F. J., & Lemons, J. E. (1996). Biomaterials science: an introduction to materials in medicine. Elsevier New York, 347-356.
Read the full article
0 notes
Text
Sanofi builds focus on rare blood disorders and cancers
Sanofi builds focus on rare blood disorders and cancers
Some of the most serious unmet patient needs today are in the field of hematology. Rare blood disorders and blood-related cancers continue to be a major focus of research as scientists look for new treatments for serious and life-threatening conditions. To help meet this need, Sanofi has significantly increased its focus on hematology, making a number of important investments to advance new therapies and improve the care of people affected by multiple myeloma, hemophilia, acquired thrombotic thrombocytopenic purpura (aTTP), cold agglutinin disease and other rare blood disorders. The company’s progress will be evident at the 60th American Society of Hematology (ASH) Annual Meeting and Exposition being held December 1 - 4 in San Diego, CA.
"Hematology has become a major focus for Sanofi, and we are very excited about the progress we are making in advancing research on a relatively broad range of potential new treatments for people with rare blood disorders and blood cancers," said Bill Sibold, Executive Vice President and Head of Sanofi Genzyme, the specialty care global business unit of Sanofi. "We look forward to sharing that progress with the hematology community at this year’s ASH meeting."
Advancements in Cancer (Hematological Malignancies)
Data presented at ASH include the latest research regarding isatuximab, an anti-CD38 monoclonal antibody in late-stage development for patients with multiple myeloma, a rare plasma cell malignancy with high unmet medical need. Isatuximab is currently being investigated in four Phase 3 clinical trials, including three company sponsored pivotal trials.
Multiple Myeloma Oral Presentations at ASH:
Results from a Phase II Study of Isatuximab As a Single Agent and in Combination with Dexamethasone in Patients with Relapsed/Refractory Multiple Myeloma (abstract 155; oral presentation on Saturday, December 1, 2018: 12:00-1:30 PM (PT))
Preliminary Results from a Phase I Study of Isatuximab (ISA) in Combination with Bortezomib, Lenalidomide, Dexamethasone (VRd), and in Patients with Newly Diagnosed Multiple Myeloma (NDMM) Non-Eligible for Transplant (abstract 595; oral presentation on Monday, December 3, 2018: 7:00-8:30 AM (PT))
Advancements in Rare Blood Disorders
Sanofi’s increased commitment to the rare blood disorders community follows the acquisition of two companies, Bioverativ and Ablynx, earlier this year. Sanofi will create a Rare Blood Disorders franchise in early 2019 that will become part of Sanofi Genzyme. In addition to two marketed therapies for the treatment of hemophilia, Sanofi’s pipeline includes novel investigational therapies in hemophilia, sickle cell disease(1), cold agglutinin disease and aTTP, a rare blood-clotting disorder.
Key Hemophilia Presentations at ASH:
BIVV001: The First Investigational Factor VIII Therapy to Break Through the VWF Ceiling in Hemophilia A, with Potential for Extended Protection for One Week or Longer (abstract 636; oral presentation on Monday, December 3, 2018: 10:30 AM-12:00 PM (PT)) (2)
ASPIRE Final Results Confirm Established Safety and Sustained Efficacy for Up to 4 Years of Treatment With rFVIIIFc in Previously Treated Subjects With Severe Hemophilia A (abstract 1192; poster on Saturday, December 1, 2018: 6:15-8:15 PM (PT)) (3)
B-YOND Final Results Confirm Established Safety, Sustained Efficacy, and Extended Dosing Interval for Up to 4 Years of Treatment With rFIXFc in Previously Treated Subjects With Severe Hemophilia B (abstract 1214; poster on Saturday, December 1, 2018: 6:15-8:15 PM (PT)) (3)
aTTP Oral Presentation at ASH:
Integrated Efficacy Results from the Phase II and Phase III Randomized Studies with Caplacizumab in Patients with Acquired Thrombotic Thrombocytopenic Purpura (abstract number 373; oral presentation on Sunday, December 2, 2018: 12:00-1:30 PM (PT))
Advancements in Rare Genetic Conditions
Sanofi Genzyme has worked in rare genetic conditions, including Gaucher disease for more than 30 years. Gaucher disease is an inherited genetic condition that can affect the blood, organs and bones. Because Gaucher disease has similar signs and symptoms of some more common hematologic malignancies, it is often misdiagnosed. Data presented at ASH includes oral eliglustat for the treatment of Gaucher disease type 1.
Gaucher Disease Presentations at ASH:
Long-Term Effects of Oral Eliglustat on Skeletal Manifestations of Gaucher Disease Type 1: Results from Four Completed Clinical Trials (abstract 2396; poster on Sunday, December 2, 6:00-8:00 PM (PT))
The Long-Term Adverse Event Profile of Oral Eliglustat for the Treatment of Gaucher Disease Type 1: Pooled Analysis of Data from 393 Patients in 4 Completed Trials (abstract 3692; poster on Monday, December 3, 6:00-8:00 PM (PT))
Isatuximab and BIVV001 are investigational agents and have not been approved by the U.S. Food and Drug Administration (FDA) or any other regulatory agency worldwide for the use under investigation.
The U.S. FDA has accepted for priority review the Biologics License Application for caplacizumab for treatment of patients 18 years of age and older experiencing an episode of aTTP. The European Commission approved caplacizumab as the first therapeutic specifically indicated for the treatment aTTP earlier this year.
Read the full article
0 notes
Text
Awful News "Bayer laying off 10 percent of its workforce"
Bayer laying off 10 percent of its workforce
In a bid to reboot its sluggish performance, the pharmaceutical and agribusiness giant has announced cuts of 10 percent of its global workforce and the disposal of underperforming divisions.
Pharmaceuticals and agribusiness giant Bayer is to cut 12,000 jobs worldwide as part of a radical restructuring plan aimed at reshaping the company by 2021. The job cuts represent 10 percent of Bayer’s global workforce.
A “significant portion” of the layoffs will be in Germany, the company said, although agreements with labor unions mean there will be no forced redundancies there until 2025. In Germany, reductions will come through natural wastage and voluntary redundancies.
As well as cutting jobs, Bayer plans a wide-ranging reorganization of the company, with major brands and even entire divisions up for disposal. In the biggest move, the company will abandon the animal health business. The sale of that division is expected to raise between €5 billion and €7 billion ($5.7–8 billion). The troubled Consumer Health division will sell key product groups, including Claritin (allergies) and Dr. Scholl (foot care).
The wave of job losses will impact all divisions. Consumer Health, whose products include aspirin, will lose 1,150 jobs. Pharmaceuticals will dispose of 1,250, including 900 in research and development. Crop Science will lose 4,100 jobs after completion of the merger with American agrichemical giant Monsanto, a recent acquisition for around $62 billion. A further 6,000 jobs will go in corporate administration.
Possibly poison
Bayer’s share price has been hit in recent weeks by fears that Bayer could end up liable for billions in compensation to those affected by glyphosate, a herbicide ingredient used by Monsanto. Last month a US court linked the substance to certain forms of cancer. The long-term implications for Bayer remain unclear.
In a conference call, CEO Werner Baumann said the restructuring had “absolutely nothing to do with” the glyphosate issue. He said the main goal was to solve long-term problems in individual divisions.
However, the scale of the reorganization suggests that Bayer is eliminating waste across its operations, in preparation for tough challenges ahead. “We’re laying the groundwork to improve Bayer’s long-term performance and earning capacity,” Baumann said. The company’s supervisory board unanimously approved the measures.
In addition to pulling out of animal health, Bayer plans to dispose of a 60 percent stake in chemical industry service provider Currenta, a joint venture with Lanxess. The sale is expected to raise around €1.5 billion. Possible buyers include former Bayer subsidiary Covestro, and specialist industry investment funds.
Poor pipeline
Analysts welcomed Bayer’s moves, suggesting it could raise up to €9 billion for investment or to pay down debt. Savings in Pharma and Consumer Health could contribute a further €1.6 billion.
Once the measures are fully implemented in 2022, Bayer hopes to save €2.6 billion annually. But costs associated with the restructuring plan could amount to €4.4 billion, as well as fourth-quarter write downs of €3.3 billion.
Bayer hopes the reorganization of its pharmaceuticals division will strengthen its unsatisfactory product pipeline, more quickly bringing new active substances to the consumer market.
The division has run into substantial difficulties in recent years, plagued with sluggish growth and diminished competitiveness. The oncology unit, for example, has made little progress in immune therapies, seen as a promising area of innovation. Restructuring the division, it is hoped, could refocus research in cutting edge fields.
In what will be a bitter blow, the company will also close a high-tech biotech facility in the city of Wuppertal, purpose-built three years ago to host blood medicine research. Competitors have taken a substantial advantage in the area; as a result, Bayer will focus exclusively on its California research facilities.
Read the full article
0 notes
Text
Validation of Assay for Simultaneous Estimation of Ebastine and Montelukast in Tablet Dosage Forms by RP-HPLC Method
INTRODUCTION
Ebastine is a second-generation H1 receptor antagonist that is indicated mainly for allergic rhinitis and chronic idiopathic urticaria. It is chemically known as 1-(4-tert-butylphenyl)-4- butan-1-one. Figure 1. It is soluble in methanol, chloroform, and dimethyl sulfoxide. Ebastine and its active metabolite is selective peripheral histamine H1 receptor antagonist. Thus it prevents the attachment of histamine on receptors and its activation (Activation of receptors of histamine on various tissues produce various allergic symptoms e.g. a Runny nose). Ebastine also has a specific inhibitory effect on Th2-type cytokine production and inhibit T cell migration and pro-inflammatory cytokine production by T cells and macrophages.
Montelukast is a leukotriene receptor antagonist (LTRA) used for the maintenance treatment of asthma and to relieve symptoms of seasonal allergies. It is chemically known as Sodium; 2- phenyl]-3- propyl] sulfanylmethyl] cyclopropyl] acetate. Figure 2. It is freely soluble in ethanol, methanol, and water. Montelukast blocks the action of leukotriene D4 on the cysteinyl leukotriene receptor CysLT1 in the lungs and bronchial tubes by binding to it. This reduces the bronchoconstriction otherwise caused by the leukotriene and results in less inflammation.
A detailed survey of the literature for Ebastine and Montelukast reveals that the available analytical methods are costly and with more retention time. Hence we developed a rapid and sensitive RP-HPLC method with UV detection (244 nm) for routine analysis of montelukast sodium and ebastine in a pharmaceutical formulation (Ebast-M). A literature review revealed few methods on method development and validation of Ebastine and Montelukast by RP-HPLC. So now the main aim is to develop a method with less run time and retention time compared to those methods.1-7
MATERIAL AND METHOD
Instruments
HPLC from Waters with model No HPLC 2965 system with Empower 2 software.
Materials
Ebastine and Montelukast (API) were received from spectrum lab, Combination Ebastine and Montelukast (EBAST M TABLET) tablets were obtained from Micro Labs, Distilled water (HPLC grade), acetonitrile, ammonium acetate buffer, methanol, Potassium dihydrogen phosphate buffer, Triethylamine, orthophosphoric acid (HPLC grade) were obtained from MERCK.
Methods
Diluent
Based upon the solubility of the drugs, diluent was selected, Methanol and Water were taken in the ratio 50:50.
Preparation of Standard Stock Solutions
Accurately Weighed and transferred 10mg and 10mg of Ebastine and Montelukast working Standards into 10ml and 10ml clean dry volumetric flasks separately, add 3/4th volume of diluent, sonicated for 30 minutes and makeup to the final volume with diluents.
Preparation of Standard Working Solutions (100% solution)
From the above each stock solution, 1 ml was pipetted out into a 10ml volumetric flask and then makeup to the final volume with diluent.
Preparation of Sample Stock Solutions
20 tablets were weighed and calculate the average weight of each tablet then the tablet powder weight equivalent to 10 mg of Ebastine and 7.5 mg of Montelukast was transferred into a 10ml volumetric flask, 7ml of diluent added and sonicated for 30 min, further the volume made up with diluent and filtered.
Preparation of Sample Working Solutions (100% solution)
From the filtered solution, 1ml was pipetted out into a 10 ml volumetric flask and made up to 10ml with diluent.
Preparation of Buffer
1ml of OPA was taken in 1000 ml volumetric flask and makeup to the mark with milli-Q water.
Preparation of Buffer: 0.01N Potassium dihydrogen orthophosphate (pH 4.8)
Accurately weighed 1.36gm of Potassium dihydrogen orthophosphate in a 1000ml of Volumetric flask add about 900ml of milli-Q water added and degas to sonicate and finally make up the volume with water the pH was adjusted to 4.8 with Orthophosphoric acid.
RESULTS AND DISCUSSION
Method Development
Table 1: Different trials were performed by changing Mobile phase and buffer
Trials
Column Used
Mobile phase
Buffer
Flow rate
Wave length
Temperature
Injection Volume
Trial: 1
Discovery 250 x 4.6 mm, 5m.
Water: Methanol (50:50)
1ml/min
244nm
25 ͦ C
10µl
Trial: 2
Discovery 250 x 4.6 mm, 5m.
Water: Acetonitrile (50:50)
Water
1ml/min
244nm
30 ͦ C
10µl
Trial: 3
Discovery 250 x 4.6 mm, 5m.
buffer: ACN (60:40)
0.1%OPA
1ml/min
244nm
30 ͦ C
10µl
Trial: 4
Discovery 250 x 4.6 mm, 5m.
buffer: Acetonitrile (60:40)
0.01N KH2PO4 (4.8) solution
1ml/min
244nm
30 ͦ C
10µl
Trial: 5
buffer: Acetonitrile (70:30A)
buffer: Acetonitrile (70:30)
0.01N KH2PO4 (4.8) solution
1ml/min
244nm
30 ͦ C
10µl
Optimized Method
Kromosil 250 x 4.6 mm, 5m.
Buffer: Acetonitrile (60:40)
0.01N KH2PO4 (4.8) solution
Diluent : Water: ACN: (50:50)
1.0ml/min
244nm
30 ͦ C
10µl
Table 2: Optimization of chromatographic conditions
Trials
Observation
Trial: 1
Ebastine peak was eluted but Montelukast peak was not eluted and peak shape also not good so further trial is carried out.
Trial: 2
Peaks were eluted but peak shape was not good and baseline disturbances hump, USP plate count were not good so further trial is carried out.
Trial: 3
Both peaks were eluted but resolution was less so further trial is carried out.
Trial: 4
Retention time is more and ebastin eluted at void range so further trial is carried out.
Trial: 5
Increasing buffer ratio montelukast retention time is more and ebastine eluted at void range so further trial is carried out.
Optimized Method
Drugs were eluted with good retention time, resolution; all the system suitable parameters like Plate count and Tailing factor were within the limits. Peak shape and retention time is good so, further process is carried out.
Method Validation
The present study was carried method was validated based on ICH (Q2B) parameters.8
The following parameters were validated for the proposed method.
System Suitability
All the system suitability parameters are within range and satisfactory as per ICH guidelines. Table 3
Discussion: According to ICH guidelines plate count should be more than 2000, tailing factor should be less than 2 and resolution must be more than 2. All the system suitable parameters were within the limits.
Discussion: Retention times of Ebastine and Montelukast were 2.447 min and 3.436 min respectively. We did not find any interfering peaks in blank and placebo at retention times of these drugs in this method. So this method was said to be specific.
Linearity
Six Linear concentrations of Ebastine (25-
150ppm) and Montelukast (20-120ppm) were prepared and injected. Regression equation of the Ebastine and Montelukast were found to be, y = 19263x +1149, and y = 19946x + 1095 and the regression coefficient was 0.999. Table 4 Figure 4 & 5
Precision
Intraday precision (Repeatability): Intraday Precision was performed and % RSD for Ebastine and Montelukast were found to be 0.2% and 0.2% respectively. Table 5
Inter-day precision: Inter-day precision was performed with 24 hrs time lag and the %RSD Obtained for Ebastine and Montelukast were 0.3% and 0.2%. Table 6
Accuracy
Three concentrations 50%, 100%, 150%, were injected in a triplicate manner and amount Recovered and % Recovery was displayed in Table 7. Figure 6-8
Robustness
Small deliberate changes in a method like Flow rate, mobile phase ratio, and temperature are made but there were no recognized change in the result and are within range as per ICH Guidelines. Table 6 Figure 9 & 10
Discussion: Robustness conditions like Flow minus (0.9ml/min), Flow plus (1.1ml/min), mobile phase minus (65B:35A), mobile phase plus (55B:45A), temperature minus (25°C) 3and temperature plus (35°C) was maintained and samples were injected in a duplicate manner. System suitability parameters were not much affected and all the parameters were passed. %RSD was within the limit.
Assay
Standard preparations are made from the API and Sample Preparations are from Formulation (EBAST M TABLET). Both sample and standards are injected six homogeneous samples. The drug in the formulation was estimated by taking the standard as the reference. The Average % assay was calculated and found to be 99.05% and 99.20% for Ebastine and Montelukast respectively. Table 7
Degradation Studies
Standards and degraded samples are injected and calculated the percentage of drug degraded in solution by applying different conditions like acid, alkali, and oxidative, photolytic, thermal and neutral analysis. Table 8 Figure 11-15
Table 3: System Suitability Studies of Ebastine and Montelukast
Property
Ebastine
Montelukast
Retention time (tR)
2.447min
3.436min
Theoretical plates (N)
8019 ± 63.48
10040 ± 63.48
Tailing factor (T)
1.37 ± 0.117
1.33 ± 0.117
Table 4: Calibration Data of Ebastine and Montelukast Method
S.No
Concentration
Ebastine (µg/ml)
Response
Concentration
Montelukast (µg/ml)
Response
1
0
0
0
0
2
25
533631
20
478732
3
50
987156
40
956442
4
75
1467357
60
1501069
5
100
1976938
80
1885033
6
125
2503069
100
2386656
7
150
3011189
120
2913242
Table 5: Repeatability results for Ebastine and Montelukast
Sl. No.
Ebastine
Montelukast
1
1958452
1868712
2
1957170
1870107
3
1952368
1865840
4
1959570
1870800
5
1953581
1865343
6
1952026
1859653
Mean
1955528
1866743
S.D.
3274.9
4115.7
%RSD
0.2
0.2
*Average of six determinations
Table 6: Inter-Day Precision Results for Ebastine and Montelukast
S. No.
Ebastine
Montelukast
1
2159276
1882066
2
2168976
1884258
3
2165538
1892454
4
2158679
1885947
5
2162743
1875128
6
2157355
1882066
Mean
2162095
1883653
S.D
4510.1
5671.0
%RSD
0.2
0.3
Table 7: Table of Accuracy
Sample
Concentration (%) (µg/ml)
Recovery (%)
Mean % Recovery
%RSD
Ebastine
50
101.07
99.93%
0.07
100
98.93
0.33
150
99.81
0.30
Montelukast
50
100.8
99.69%
0.08
100
99.63
0.74
150
98.64
0.52
Table 8: Robustness Data of Ebastine and Montelukast
S. No
Robustness condition
Ebastine
%RSD
Montelukast
%RSD
1
Flow minus (0.9ml/min)
0.1
0.2
2
Flow Plus (1.1ml/min)
0.4
0.5
3
Mobile phase minus (65:35)
0.3
0.3
4
Mobile phase Plus (55:45)
0.2
0.1
5
Temperature minus (250c)
0.2
0.2
6
Temperature Plus (300c)
0.3
0.1
Table 9: Assay of Tablet
S. No.
Ebastine %Assay
Montelukast % Assay
1
99.15
98.91
2
99.23
99.19
3
99.00
98.10
4
99.26
99.84
5
98.97
99.46
6
98.67
99.70
AVG
99.05
99.20
S.D
0.2184
0.6339
% RSD
0.2
0.64
Table 10: Different Types of Degradation Studies
Types of Degradation
EBASTINE
Area
%
Recovered
%
Degraded
Acid
1800991
95.56
4.44
Base
1833505
97.28
2.72
Peroxide
1853657
98.35
1.65
Thermal
1871146
99.28
0.72
UV
1868750
99.15
0.85
Water
1867367
99.08
0.92
MONTELUKAST
Acid
1867882
95.28
4.72
Base
1910567
97.45
2.55
Peroxide
1930866
98.49
1.51
Thermal
1950620
99.50
0.50
UV
1951371
99.54
0.46
Water
1947871
99.36
0.64
Figure 1: Structure of Ebastine
Figure 2: Structure of Montelukast
Figure 3: Typical chromatogram of Ebastine and Montelukast
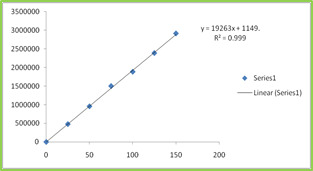
Figure 4: Calibration curve of Ebastine
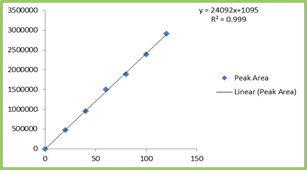
Figure 5: Calibration curve of Montelukast
Figure 6: Accuracy 50% Chromatogramof Ebastine and Montelukast
Figure 7: Accuracy 100% Chromatogram of Ebastine and Montelukast
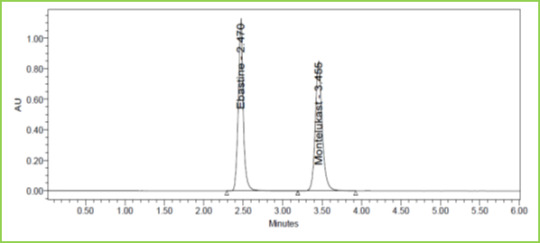
Figure 8: Accuracy 150% Chromatogram of Ebastine and Montelukast
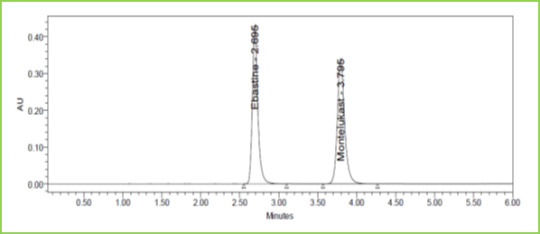
Figure 9: Flow minus Chromatogram of Ebastine and Montelukast
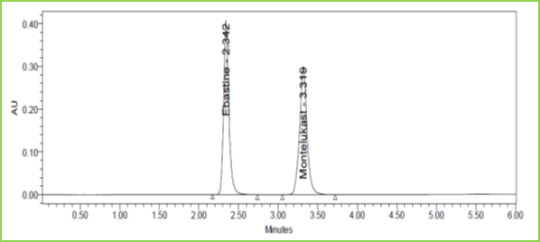
Figure 10: Flow plus Chromatogram of Ebastine and Montelukast
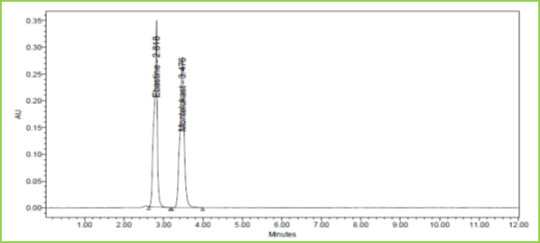
Figure 11: Chromatogram showing Acid degradation
CONCLUSION
A new method was established for simultaneous estimation of Ebastine and Montelukast by RP-HPLC method. The proposed HPLC method was found to be simple, specific, precise, accurate, rapid and economical for simultaneous estimation of Ebastine and Montelukast in the pharmaceutical dosage form. The developed method was validated in terms of accuracy, precision, linearity, robustness, and ruggedness, and results will be validated statistically according to ICH guidelines. The Sample recoveries in all formulations were in good agreement with their respective label claims. Hence the suggested RP-HPLC method can be used for routine analysis of Ebastine and Montelukast in API and Pharmaceutical dosage form.
ACKNOWLEDGMENTS
I am very much thankful to the management of Seven Hills College of Pharmacy for providing the necessary facilities to carry out the research work and for their encouragement and support.
REFERENCES
Rana, N. S., Rajesh, K. S., Patel, N. N., Patel, P. R., Limbachiya, U., & Pasha, T. Y. (2013). Development and validation of an RP-HPLC method for the simultaneous estimation of montelukast sodium and ebastine in tablet dosage form. Indian Journal of Pharmaceutical Sciences, 75(5), 599.PMid:24403662, PMCid:PMC3877523
Singh, R. M., Saini, P. K., Mathur, S. C., Singh, G. N., & Lal, B. (2010). Development and validation of an RP-HPLC method for estimation of montelukast sodium in bulk and in tablet dosage form. Indian Journal of Pharmaceutical Sciences, 72(2), 235.https://doi.org/10.4103/0250-474X.65023 ,PMid:20838530 , PMCid:PMC2929785
Yadav, O. M., & Jain, H. K. (2014). RP-HPLC Method Development and Validation for Simultaneous Estimation of Phenylephrine Hydrochloride and Ebastine in Tablet Dosage Form. International Journal of Pharmacy and Pharmaceutical Sciences, 6(8), 466-470.
Savsani, J. J., Goti, P. P., & Patel, P. B. (2012). Development and validation of simultaneous equation method for estimation of ebastine and montelukast sodium in combined tablet dosage form. Der Pharmacia Sinica, 3(6), 690-698.
Shrikrishna, B., & Nisharani, R. (2015). Analytical Method Development and Validation for Simultaneous Estimation of Montelukast and Ebastine by HPLC. Research Journal of Pharmacy and Technology, 8(1), 1. https://doi.org/10.5958/0974-360X.2015.00001.3
Singh, K., Bagga, P., Shakya, P., Kumar, A., Khalid, M., Akhtar, J., & Arif, M. (2015). Validated UV Spectroscopic Method for Estimation of Montelukast Sodium. International Journal of Pharmaceutical Sciences and Research, 6(11), 4728-4732.
Thakor K. A, Pasha, T. Y., Patel P. U., Chauhan R. J., Patel N. H. (2014). Development and validation of analytical method for simultaneous estimation of ebastine and phenylephrine hydrochloride in tablet dosage form. International Bulletin of Drug Research, 4(7): 16-40, 2014.
8. ICH Harmonised Tripartite Guideline, validation of analytical procedures: Text methodology, Q2 (R1) (2005). International Conference on Harmonization, Geneva, pp: 1-13.
Read the full article
0 notes
Text
Intestinal Parasitic Infection and Nutritional Status among Urban and Rural Population of Khurja, Bulandshahr (U.P.)
INTRODUCTION
Human intestinal parasites occur throughout the world but it is in the west tropics and sub-tropics where they are found in their greatest numbers. A basic requirement for the continued survival of these organisms is an inadequate and unhygienic method of disposal of faecal material. The intestinal parasitic infections caused by intestinal parasites are among the most prevalent infections in human in developing countries. Intestinal parasitic infections such as amoebiasis, ascariasis, hookworm infection and trichiuriasis are among the ten most common infections in the world . Human intestinal parasites can be present in any disease, in any person, at any age. People with intestinal parasitic infections are usually under nourished and weak, infected with viral, fungal, or bacteria, and have various types of chemical and metal poisoning. Intestinal parasites cause a significant morbidity and mortality in endemic countries. These infections are the most prevalent in tropical and sub – tropical regions of the developing world where adequate water, sanitation facilities and poor economic conditions are lacking. The worldwide prevalence of intestinal parasites is estimated in more than 3.5 billion with around 4.5 million clinical cases. . It is observed that about 60 – 80 percent population of certain areas of West Bengal, Uttar Pradesh, Bihar, Orissa, Punjab, East Coast of Tamil Nadu and Andhra Pradesh is infected with parasites . Intestinal parasitoses are common both in general population and in people residing in institutions in tropical and sub – tropical regions. The conditions required for transmission and aqusion of intestinal parasitism are favored in institutions where large number grouped together for a long period of time and poor sanitary conditions prevail. This is evidenced by studies on the prevalence of intestinal parasites in school, day care centers and institutions. Local conditions such as quality of domestic and village infrastructure, economic, occupation and social factors such as education influence the risk of infections, diseases transmission and associated morbidity and mortality. The objective of this study was to perform an epidemiological survey to determine the prevalence of intestinal parasitic infections in the populations of Khurja, Bulandshahr.
MATERIAL AND METHOD
The present study was conducted on human intestinal parasitic patients and few healthy subjects as control. In this study, a survey was carried out for human parasitic diseases, from rural and urban populations of Khurja, Bulandshahr for two years from 2009 to 2011. For this study, an interview technique was performed to collect the information of subjects regarding their age, sex and family background. For the present study, a total of 223, samples of stool for both rural and urban populations were collected for microscopic investigations in laboratory. The Simple Smear in Saline method was used to determine the stool samples. The persons having any cyst/ova/trophozoit/whole parasite were treated as parasitic positive patients. During the Demographic study of persons, the age group, sex, socio-economic and literacy status were included in this study. The Chi –Squared tests were performed to the test for an association between all possible pairs of parasitic infections and between the genders of each age group. The calculated χ2 value was more than P – value (at 0.05 level).
RESULTS AND DISCUSSION
Overall 223, stool samples were examined by Simple Smear in Saline method on the population of Khurja, Bulandshahr. The age combination shows that 52 (23.31%) persons were in 0-15 age group and 91 (40.80%) in 15-35 age group while, 80 (35.87%) were above the age of 35 years. The sex based distribution shows that out of 223 samples, the 120 (53.81%) were collected from males and 103 (46.18%) from female. According to socio-economic status 71 (50.70%) to low (5001 to 15000), 112 (17.85%) to medium (15001 to 25000) and 40 (12.5%) persons to high (
Read the full article
0 notes
Text
Computational Drug Designing of Anticancer Drugs
INTRODUCTION
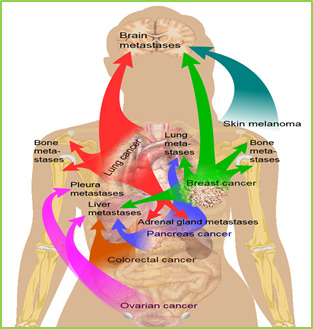
Figure 1: Cancer metastasis (spread) to different parts of the body
Cancer is one of the hazardous and convoluted disease. Cancer is the main source of overall demise even after a ton of headway as far as determination and treatment in the previous few years. It is anticipated that the malignancy passing rate may achieve 13.2 million by the year 2030.Cancer includes hereditary and epigenetic reconstructing of typical body cell prompting destructive cell which at last outcomes in everlasting status and uncontrolled division.
The uncontrolled cell divisions increments with increment in tumor mass bringing about metastasis and demise of entire living being because of organ failure. Metastases is one of the greatest test to medicinal administration of growth and one of the significant reason for death in malignancy patients.
Identification of the component that prompts tumorigenesis and malignancy movement helps in discovering more strong therapeutics and enhanced diagnostics. Over the previous couple of decades several growth target (proteins, compounds or receptors) has been found and therapeutics are composed in view of these targets.1The current medication accessible for disease treatment are extremely costly, profoundly inadequate, nonspecific and has number of side effects.2
Computational drug design, set up well ordered throughout the most recent couple of decades, makes it conceivable to dispose of a significant number of the previously mentioned issues. Utilizing a wide range of calculations, approximations of restricting free vitality of synthetic mixes to a sub-atomic target can be produced in silico in a quick and exceptionally shabby path, with no requirement for physical accessibility of those compounds in this progression. PC helped sedate outline in this way permits to radically accelerating the errand of growing new medications, firmly diminishes costs and empowers the quick testing of new, yet non-blended, classes of compounds. Besides, it can likewise be utilized to anticipate other substance properties of atoms, similar to their ingestion, circulation, digestion, and excretion, and along these lines accelerate tranquilize improvement by either evacuating compounds with undesired properties or by streamlining properties of found hits. Keeping in mind the end goal to make utilization of the considerable number of focal points of present day PC supported medication outline, a plenty of various calculations and readiness steps is vital, which have be to used together in enormous computational pipelines.3
One of the regions of utilization of computational drug designing is quantitative structure action (QSAR) demonstrating. A wide range of relapse and arrangement models, and additionally the info age, information administration, include choice and model approval systems under one basic structure, so the greater part of the methodology are anything but difficult to use in blend are depicted which are rapidly extensible and adaptably usable. For the field of structure-based medication outline, a quick receptor-ligand scoring capacity, a docking calculation and a three-dimensional target-particular rescoring approach was produced by the researchers. Besides, to take into account advancement of restricting free vitality gauges got by docking, another receptor-ligand rescoring strategy was executed. It utilizes the three-dimensional data (i.e., the purported postures depicting the putative ligands inside the coupling pocket) produced by docking and test restricting free vitality estimations for different mixes so as to reestablish the docking postures. In this manner, this approach, as opposed to every single other one known, considers receptor-ligand associations, their three-dimensional areas and their objective particular significance.4
The overview of entire framework is called CADDSuite (Computer-Aided Drug Design Suite). This framework contains all the algorithms and a high number of auxiliary tools, e.g. for preparation or analysis purposes. An introduction to biochemical and computational background is also described.
Drug Discovery and Development Process
Overview of Drug Design
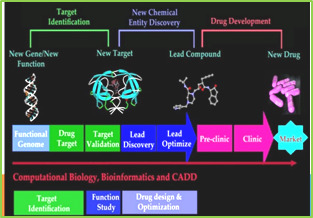
Figure 2: Overview of usual process of drug development
Drug discovery and advancement is a complex, lengthy, time expending and exceptionally costly process. It includes the cooperation of different fields, for example, medicinal chemistry, pharmacology, clinical research, drug metabolism, process chemistry.....etc.
Additionally combinatorial chemistry, high throughput screening and molecular modelling assumes a fundamental part in current drug discovery process.
It takes around 7-12 years and $ 800 million to $1.8 billion to bring new lead from drug revelation to market. Initially 1,00,000 applicant compounds, hundred of preclinical animal testing and clinical trails on thousand of volunteers and patients is conveyed to recognize a solitary showcased drug. The process from the ID of new medication to the promoting is alluded as pipeline which includes following real advances.5
Disease selection
Target identification
Lead identification
Lead optimization
Preclinical trials
Clinical trials
Computational Background
Overview of Computer-Aided Drug Design
Computational drug designing is the advanced process of drug discovery and development of new drugs.with the advent of this computational drug designing many of the problems so associated with the rational drug designing can be overcome.This technology has helped to reduce the cost and amount of time spend to develop a new drug.
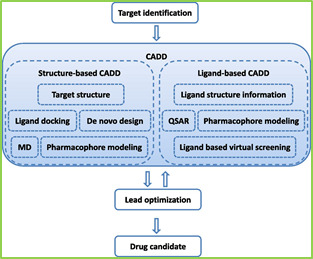
Figure 3: Flow chart of CADD in drug discovery/design
Furthermore, computer-aided drug design may also establish molecules as promising drug candidates that would never have been tested without computer-based methods, due to either the huge search-space or their initial unavailability in synthesized form. A special case of the latter reason is the in silico construction of new molecules, i.e. compounds that have not been observed in nature but were constructed on a computer manually or by an algorithm designed for this purpose. In essence, computer-aided drug design approaches try to predict properties and actions of chemical compounds by a variety of techniques, so that molecules that are unlikely to experience the desired effect on the chosen molecular target can be cast aside. Examples of such molecular properties are absorption, distribution, metabolism, excretion and toxicity (ADMET). The expected effect on the molecular target, on the other hand, is usually evaluated by a prediction of the binding free energy (or binding affinity) of the compound to the target structure. Computer-aided drug design can be divided into two major categories: ligand-based drug design and structure-based drug design.
Ligand-Based Computer-Aided Drug Design:
The ligand-based computer aided drug design (LB-CADD) approach includes the investigation of ligands known to interface with an objective of intrigue. These techniques utilize an arrangement of reference structures gathered from Compound known to interface with the objective of intrigue and examine their 2D or 3D structures. The general objective is to speak to these mixes in such a way that the physicochemical properties most important for their coveted communications are held, though superfluous data not pertinent to the communications is disposed of. It is viewed as a backhanded way to deal with medicate disclosure in that it doesn't require learning of the structure of the objective of interest. The two crucial methodologies of LB-CADD are (1) determination of mixes in view of substance similitude to known actives utilizing some similitude measure or (2) the development of a QSAR demonstrate that predicts biologic movement from synthetic structure. The contrast between the two methodologies is that the last weights the highlights of the compound structure as indicated by their effect on the biologic movement of intrigue, while the previous does not. The strategies are connected for in silico screening for novel compound having the biologic action of intrigue, hit-to-lead and prompt medication advancement, and additionally for the enhancement of DMPK/ADMET properties.LB-CADD depends on the Similar Property, which expresses that atoms that are fundamentally comparable are probably going to have comparable properties. LB-CADD approaches rather than SB-CADD methodologies can likewise be connected when the structure of the biologic target is obscure. Furthermore, dynamic mixes recognized by ligand-based virtual high-throughput screening (LB-vHTS) strategies are regularly more powerful than those recognized in SB-vHTS
Structure based Computer-Aided Drug Design:
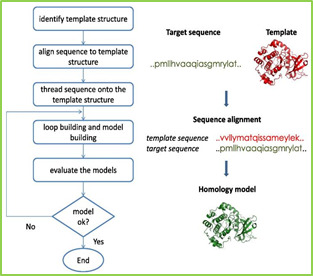
Figure-4: Flow chart of Structural based Computer Aided Drug Design (Homology model Building Process)
Structure-based computer aided drug design utilizes the three-dimensional structure of the objective of enthusiasm for request to discover aggravates that reasonable tie to its coupling pocket and could subsequently be great drug applicants. The three-dimensional structure can be acquired by either X-ray protein crystallography, atomic attractive reverberation (NMR), or by homology modelling. The last method utilizes the structure of a homologous protein that has been dictated by one of the previous techniques. Calculations for the field of structure-based medication configuration fundamentally comprise of receptor ligand docking and rescoring approaches.
The objective of receptor-ligand docking is to anticipate the binding of a ligand in the coupling pocket of a receptor, given just the 3D directions of the last mentioned and the topology (or information adaptation) of the previous. In this way, docking approaches more often than not comprise of a scoring capacity that assesses the collaboration vitality of each (moderate) posture and a calculation that creates a wide range of poses to be assessed by the scoring capacity. Scoring capacities can for the most part be isolated into learning based and observational ones. While the previous utilize a reversal of the Boltzmann factor to compute scores from the recurrence of various perceptions, the last utilize various (regularly physically inspired) terms whose coefficients are advanced utilizing a particular informational index with known binding free energies.
Chemical Classification of Anticancer Drugs
The drugs used in the treatment of cancer are classified into different classes which are described as follows

Figure. 5: Chemical classification of Anti-cancer Agent

Figure 6: chemical structure of alkylating agents
Alkylating Agents
Nitrogen mustard: eg Mustine HCl, Mechlorethamine
Ethylene mine: eg Thiotepa
Alkyl sulphonate: eg Busulfhan
Nitrosourea: eg Carmustine,Lomustine
Triazine Dacarbazine
Platinum complex: eg cisplastin, carboplatin
Antimetabolites:
Folate antagonist: eg Methotrexate
Antipurine: eg 6MP,Pentostatin
Antipurine: 5 fluorouracil,Gemcitabine
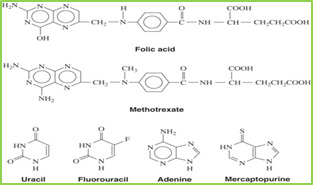
Figure 7: chemical structure of antimetabolites
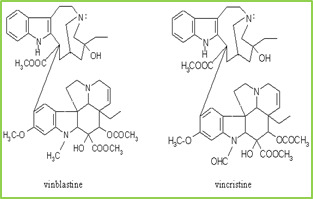
Figure 8: chemical structure of vinblastine and vincristine

Figure 9: chemical structure of paclitaxel and Docetaxel
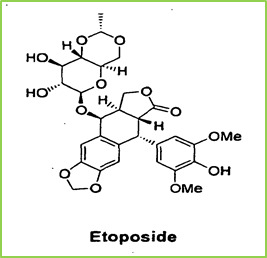
Figure 10: chemical structure of Etoposide
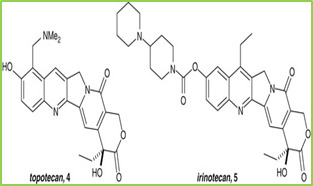
Figure 11: chemical structure of Topotecan and Irinotecan
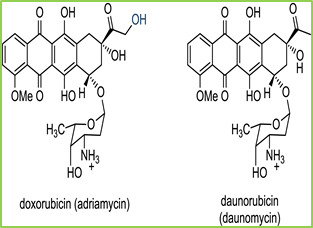
Figure 12: chemical structure of Doxorubicin and Daunorubicin

Figure 13: chemical structure of Hydroxyurea
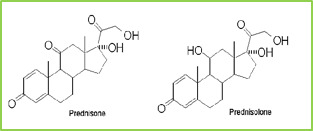
Figure 14: chemical structure of Prednisone and prednisolone
TARGETS FOR CANCER
Since convectional chemotherapy isn't particular for malignancy cells prompting harmful reactions there is a requirement for novel specialists with high review antitumour specificity.The significant essential to grow such medications is to comprehend the objectives that these operators should attack. In late years a number of promising new anticancer medications have been produced which target intracellular pathways or extracellular cell molecules.so a portion of the major targets are recorded underneath.
Topoisomerase as anticancer target

The expression "chemotherapy" was initially instituted by the renowned German scientist Paul Ehrlich in the mid 1900s. It was for the most part characterized as the approach of utilizing chemical to treat illnesses. In any case, these days this term as a rule alludes to treatment with the chemicals that kill the quickly dividing cells. Obviously, some non-malignancy cells likewise partitioned rather quickly, e.g., cells in bone marrow, stomach related tract, and hair follicles, and subsequently chemotherapy are for the most part considered non-particular and are called "cytotoxic." Common chemotherapeutic specialists incorporate alkylating operators, antimetabolites, antimicrotubule agents, cytotoxic anti-infection agents, and topoisomerase inhibitors or toxic substances. A brilliant late verifiable record of the improvement of tumor chemotherapy is accessible. In this segment, we will center around topoisomerase harms since they are among the best and most generally utilized anticancer medications. For instance, doxorubicin (Adriamycin), an outstanding Top2 poison, is as yet dynamic as a first-line treatment for breast cancer, bone and soft tissue sarcomas, anaplastic thyroid tumor, bladder growth, numerous myeloma, and Hodgkin's and non-Hodgkin's lymphomas, and so on. Topoisomerases settle topological issues of DNA two fold helices by giving the broken strand a chance to pivot around the in place strand (named Type IB, or Top1 for people), passing one strand (named Type IA, or Top3 for people) or duplex (named Type IIA, or Top2 for people) from a similar DNA atom through the single or two fold stranded break they produced on another duplex. Up until now, there is no medication focusing on Type IA topoisomerase. All FDA-endorsed drugs focusing on Type IB topoisomerase are camptothecin subsidiaries. Topoisomerase poison act by catching the chemical as a fruitless protein DNA-tranquilize "cleavage complex" (DNA is cut in this structure), which adequately changes over the compound into a cell harm lastly prompts apoptosis of the tumor cells.

Figure 15: Molecular graphics of the snapshot of the Top2β-DNA-VP16 (etoposide) ternary complex in the end of the explicit-solvent molecular dynamics simulation
The X-beam structures of human DNA topoisomerase in complex with DNA and the camptothecin simple were resolved in 2002. The synthetic assorted variety of Top2 harms is substantially higher. There are two isoforms of Top2: Top2α and Top2β. The structure of Top2α in complex with DNA, however without tranquilize, was distributed in 2012.The structures of Top2β in complex with DNA and medications were resolved as of late. Restraint of Top2β has been perceived to be in charge of the cardiotoxicity of a few medications, e.g., doxorubicin, and along these lines has been considered as a "anti-target" for planning new topoisomerase harms.
To segregate the drug binding modes in Top2α and Top2β, we initially connected sub-atomic docking with our recently created scoring capacity to assess the coupling methods of some Top2 drugs, in particular, VP-16 (etoposide), m-AMSA and mitoxantrone. Our docking computations very much recreated the crystallographic restricting method of VP-16 of every a ternary complex of Top2β; with a root-mean-square deviation of just 0.65 Å. Sub-atomic progression reproduction of Top2β in complex with VP-16 likewise affirmed the crystallographic restricting mode. Interestingly, the compliances of Arg503 of Top2β in complex with m-AMSA and mitoxantrone from the atomic progression reproductions goes astray from their unique crystallographic adaptations, showing an unwinding procedure from the adaptations decided with the medication substitution strategy for setting up the holo protein precious stones. The coupling method of VP-16 in the cleavage complex of Top2α was controlled by the joined utilization of homology displaying, docking, and sub-atomic progression reproductions, which fell inside a comparative restricting pocket of Top2β cleavage complex. The dynamic data of Top2α and Top2β may encourage more productive planning toward Top2α-particular medications.
The binding modes in the molecular dynamics simulations are consistent with the crystallographic binding mode of this ternary complex.
Mutant RAS protein inhibitors

In around 20– 30% of every single human tumor, transformations of proteins in the Ras (curtailed from "rodent sarcoma") family are much of the time watched. Ras proteins are little GTPases, and are known to shape nanoclusters on films. The Nano space area and film introduction of Ras could create isoform diversity. Creating helpful operators that can return the abnormal flagging caused by the mutant Ras proteins is considered as a powerful approach for malignancy chemotherapy.
Figure 16: The X-ray crystallographic binding mode of N-{1- piperidin-4-yl}-4-sulfanylbutanamide (designated as 6) bound with K-Ras G12C mutant. The hydrogen bond between 6 and Gln61 is shown as a thin yellow line
In the previous decades, we have exceptional advances for the basic and dynamical portrayals of the Ras proteins, particularly with molecular dynamic simulation in unequivocal dissolvable and express lipid condition. For instance, it was discovered in light of molecular dynamics simulation that the K-Ras (found by Werner Kirsten) are more adaptable than N-(acronym of neuroblastoma) and H-Ras (found by Jennifer Harvey). By one next to the other examination of FRET tests and molecular dynamics simulation, a novel switch locale of H-Ras was recognized. The multi-boundary crossing conformational changes of proto-oncogenic H-Ras between GDP-bound and GTP-bound states were effectively portrayed with quickened sub-atomic flow reenactments. The wild-type K-Ras and mutant H-Ras A59G were observed to be characteristically more unique than the wild-type H-Ras. The association, elements, and isolation of Ras nanoclusters in film areas have been described by coarse-grained semi-atomistic sub-atomic progression reproductions. There are also energizing drug discovery comes about on the Ras proteins.
For instance, staurosporines have been found to disturb phosphatidylserine trafficking and to mis-restrict Ras proteins. Andrographolide subsidiaries have been found to hinder the GDP/GTP nucleotide trade and to revoke oncogenic Ras work. Recently, the coupling hotspots on K-Ras were related to the test based sub-atomic flow reenactments, which encourage denovo drug design of new little particles.
Kinase Inhibitor

Figure 17: The X-ray crystallographic binding mode of skepinone-L bound with p38α mitogen-activated protein kinase (MAPK). The PDB accession number is 3QUE.The hydrogen bonds between skepinone and the backbone nitrogen and oxygen atoms of Gly100 are shown as thin yellow linesSupported by the 2001 FDA-endorsement of the primary kinase inhibitor, imatinib (or Gleevec), a point of reference work of disease therapeutics, kinases have turned out to be a standout amongst the most seriously sought after focuses in late pharmaceutical inquires about. Until July 2015, a sum of 28 little particle kinase inhibitors have been endorsed. In spite of the noteworthy accomplishments in the improvement of kinase inhibitors, drug resistance is as yet one of the focal issues for growing far superior therapeutics, and the unconstrained mutations in the ATP-binding space of the kinases are a standout amongst the most imperative causes.
As of late, long (18.5 μs) express solvent thermodynamics combination counts for the protection causing changes of p38α MAPK were led to accomplish great relationships amongst's tentatively and computationally decided binding free energies.
This p38α MAP kinase was known to have an elective restricting site close to Phe169 after authoritative of diaryl urea inhibitor BIRB796, and unequivocal solvent molecular dynamics simulations could over and again recognize this mysterious binding site. A computational strategy, SCR, which unequivocally assesses receptor adaptability with the rotamer library and with full atomic points of interest, was proposed to plan kinase inhibitors. This strategy could duplicate the known binding methods of the benchmarked kinase inhibitors.
It is fairly normal that aggravates that objective kinases will tie to a group of kinases, rather than one particular kinase. Knowing the range of the kinase restricting profile is likewise vital for deducing the most reasonable utilizations of the new mixes. For this situation, not just one single protein ought to be considered as the objective of medication outline. As a rule, distinguishing proof of conceivable biomolecular focuses (off-targets) of little chemical particles is a vital advance for disentangling the hidden reasons for their activities at the sub-atomic level. To this end, we have built a web server, ID Target (http://idtarget.rcas.sinica.edu.tw) that can anticipate conceivable binding targets of a little synthetic particle by means of a divide-and-conquer docking approach, in mix with our as of late created scoring capacities that depend on vigorous relapse examination and quantum compound charge models. It was exhibited that ID Target can recognize kinase inhibitors. Scientists likewise demonstrate that ID Target can repeat known off-targets of drugs or drug like compounds, and it is conceivable that the proposed new targets could be abused for advance applications.
HDAC inhibitor
Figure 18: The binding pose of the ligand N-(4-aminobiphenyl-3-yl) benzamide (designated as LLX) in the original crystal structure of HDAC/inhibitor complex (yellow), the pose with local optimization (cyan), and the pose predicted with AutoDock4 docking with the scoring function AutoDock4RAP(pink). The root-mean-squared deviations of the two predicted ligand poses from the crystal pose are 0.315 Å and 0.277 Å, respectivelyDouble activity Compound have a place with the least complex class of "designed multiple ligands" (DMLs), which are single compounds intended to at the same time regulate numerous objectives. As a result of the engineered possibility and the medication resemblance limitations, the majority of DMLs are double activity Compounds. In the previous decade, outlining double activity Compounds has turned into a developing worldview for drug discovery. In this survey, we will center around double activity intensifies that fuse some portion of the inhibitors of histone deacetylase (HDAC) as the substance moieties.
Deacetylation of the lysine buildups of histones by HDAC brings about a profoundly minimal condition of chromatin, which makes those areas of the chromosomes have a transcriptionally inert state. HDAC overexpression has been found in an assortment of human malignancies, including myeloid neoplasia and solid tumors. The relationship of HDACs with oncogenic DNA-restricting combination proteins and other harsh interpretation factors constitutively smothers particular tumor silencer qualities. Subsequently, HDACs have been considered as a critical class of targets for disease treatment. A few HDAC inhibitors (HDACis) are as of now under clinical trials on either monotherapy or mix treatment for malignancy treatment. It isn't astonishing to see that the idea of DMLs has been connected to plan new HDAC's by consolidating other dynamic operators focusing on inosine monophosphate dehydrogenase, atomic vitamin D receptor, tyrosine kinase receptor or topoisomerase II 24 in growing new therapeutics for tumor medications.
A moderately less verifiable truth is that statins are likewise HDAC inhibitors. Statins, for example, lovastatin and atorvastatin, are best known to diminish serum cholesterol levels through focused restraint at 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMG-CoA reductase, or HMGR). The HMGR inhibitors (HMGRis) are viably used to diminish the rate of cardiovascular and cerebrovascular issue and to forestall cardiovascular disease (CVD). Statins have a built up record of security and adequacy in human CVD counteractive action. In spite of the fact that statins have as of late been appeared to be viable for tumor avoidance in observational, preclinical, and certain randomized controlled investigations, the fundamental molecular mechanism is as yet subtle. Our examinations showed that statins specifically follow up on HDACs as inhibitors, which gives an epigenetic component to tumor aversion and malignancy treatment.
It has been accounted for that the joined utilization of anticancer specialists with statins may decrease reactions to accomplish better treatment of growths. Besides, the in vitro try utilizing a mix of HDACi and HMGRi has demonstrated some level of synergism for the enlistment of apoptosis of HeLa cells. The basic synergistic system has been proposed as takes after: the down-direction of GGTase-I β subunit, caused by HDACi (TSA in that review), upgrades the exhaustion of mevastatin-prompted geranylgeranylated RhoA.
Given the previously mentioned confirm, it is possible that simultaneous hindrance of HDAC and HMGR would be a promising methodology for growth treatment. In any case, there will be some known disadvantages if multi-component drug cocktail are adopted for example, complex pharmacokinetics, unpredictable drug to drug interactions and formulation issues because of various solvency of individual drugs.
Scientists have in this manner set out to outline a progression of double activity Compounds to target histone deacetylase (HDAC) and 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGR) correspondingly. These compounds indicated powerful inhibitory exercises against HDACs and HMGR with IC50 esteems in the Nano molar range. These compounds viably diminished the HMGR action and in addition advanced the acetylation of histone and tubulin in malignant cells, however were not dangerous to typical mouse fibroblast cells and human fibroblast cells.10
Current Status
Figure 19: Structure of tubulin along with its inhibitors
Earlier the development of anticancer agents used to be a lengthy, time consuming and expensive process but thanks to computational drug designing which has lower the investment on technologies, time required and resources. Large amount of information available about the small molecules, target structure and advancement in genomics and proteomics has helped in application of computational drug designing every step of drug discovery and development .The 3D structure of target and chemical compounds have increased the affinity towards the target with the aid of computational methods. The protein structure so determined by X-ray crystallography and NMR technique can be used as drug target and help to design target inhibitors which can fit in the binding pocket of the protein. some of the examples of successful application of computational drug designing are listed below.11
Figure 20: Role of p53 unregulated modular of apoptosis(PUMA) in apoptosis
Tubulin is a protein which polymerised to form microtubule which is a component of eukaryotic cytoskeleton. It plays an important role in cell proliferation and division and also a major target in anticancer therapy. Tubulin inhibitors are developed by ligand based drug designing in order to prevent the cancer cell division.
Prior the improvement of anticancer agents used to be an extensive, tedious and costly process however on account of computational drug designing which has bring down the venture on innovations, time required and resources. Large measure of data accessible about the small molecules, target structure and progression in genomics and proteomics has helped in use of computational drug designing in each progression of drug discovery and advancement .The 3D structure of target and synthetic compounds have expanded the proclivity towards the objective with the guide of computational methods. The protein structure so dictated by X-RAY crystallography and NMR strategy can be utilized as medication target and help to configuration target inhibitors which can fit in the coupling pocket of the protein. some of the cases of fruitful use of computational medication planning are recorded below.
Tubulin is a protein which polymerised to shape microtubule which is a segment of eukaryotic cytoskeleton. It assumes an imperative part in cell multiplication and division and furthermore a noteworthy focus in anticancer therapy. Tubulin inhibitors are produced by ligand based drug designing so as to stop the cancer cell division.
P53 unregulated modulator of apoptosis (PUMA) is an apoptotic protein having a place with the group of Bcl-2 protein which is managed by tumor silencer p53 and causes apoptosis. PUMA inhibitors are created by structure based pharmacophore modelling keeping in mind the end goal to represses apoptosis.
Table 1: selected inhibitors developed with computational designing
Compond Name
Therapeutic Area
Function
Approvals
Gefitinib
NSCLC
EGFR kinase inhibitor
2003
Erlotinib
NSCLC
pancreatic cancer
EGFR kinase inhibitor
2005
Sorafenib
Renal cancer
Liver cancer
Thyroid cancer
VEGFR kinase inhibitor
2005
Lapatinib
ERBB2- positive breast cancer
EGFR inhibitor
2007
Abiraterone
Metastatic castration -resistant prostrate cancer or hormone refractory prostrate cancer
Androgen synthesis inhibitor
2011
Crizotinib
NSCLC
ALK inhibitor
2011
Computational drug designing has generated large amount of new anticancer drugs and are a milestone in drug discovery.12
FUTURE PERSPECTIVES
With the expansion in the accumulation of data about the little atoms and biomolecular structure utilizing structure and ligand based approaches, there is an improvement in computational medication planning of anticancer drugs. In future better comprehension about etiology of infections would help in the revelation of new medication targets and furthermore blend of targets. The whole natural framework can't be recreated on the computer so endeavors ought to be made to include all the conceivable parameters. Unavailability of reliable experimental information and confinements in toxicity expectation display are a portion of the difficulties looking by computational drug designing. Advanced toxicity forecast model ought to be produced so as to evaluate the poisonous quality of drug in kidney, liver, lung, heart and other organs. Mechanism of diseases, genomics and proteomics, new drug targets, natural leads, physiochemical properties and so on should be recognized later on so as to get huge accomplishment in growth treatment.13
CONCLUSION
Computational drug designing techniques have extraordinary potential in drug discovery especially in lead distinguishing proof and lead optimization. It is an advanced, convenient and quickened strategy for sedate revelation and development. Numerous of therapeutic agents have been computationally intended to treat cancer, so computational drug discovery and development holds a great promise for future growth of anticancer drugs.
With the regularly expanding accumulation of biomolecular structures, constant upgrade of computational power, and enhanced correctnesses in displaying the sub-atomic communications at the nuclear level, it is expected that calculation will play a significantly more vital part in the drug discovery process sooner rather than later. Better understandings of the etiology of illnesses with the assistance of biological system and frameworks pharmacology additionally prompt distinguishing proof of new medication targets and furthermore imaginative combination of drug targets for designing new drugs all the more viably.
REFERENCES
Carels, N., Tilli, T., & Tuszynski, J. A. (2015). A computational strategy to select optimized protein targets for drug development toward the control of cancer diseases. PloS one, 10(1), e0115054. https://doi.org/10.1371/journal.pone.0115054
Ferreira AK, Kawamura B, Jorge SD, de Azevedo RA, Zaim MH, et al. (2017) Developing Novel Anticancer Drug Candidates Regarding the Integration of Three Main Knowledge Fields: Computer-Aided Drug Design, Chemical Synthesis, and Pharmacological Evaluation. J Drug Des Res, 4(2): 1035.
Sugrue, M. F. (2000). Pharmacological and ocular hypotensive properties of topical carbonic anhydrase inhibitors. Progress in retinal and eye research, 19(1), 87-112. https://doi.org/10.1016/S1350-9462(99)00006-3
Monroe, D. (2007). Looking for chinks in the armor of bacterial biofilms. PLoS biology, 5(11), e307. https://doi.org/10.1371/journal.pbio.0050307 , PMid:18001153, PMCid:PMC2071939
Gao, C. (2016). Computer-aided drug design approaches in developing anti-cancer inhibitors.
Sliwoski, G., Kothiwale, S., Meiler, J., & Lowe, E. W. (2014). Computationalmethods in drug discovery. Pharmacological reviews, 66(1), 334-395. https://doi.org/10.1124/pr.112.007336 , PMid:24381236, PMCid:PMC3880464
Muegge, I., & Martin, Y. C. (1999). A general and fast scoring function for protein− ligand interactions: a simplified potential approach. Journal of medicinal chemistry, 42(5), 791-804. https://doi.org/10.1021/jm980536j, PMid:10072678
Böhm, H. J. (1994). The development of a simple empirical scoring function to estimate the binding constant for a protein-ligand complex of known three-dimensional structure. Journal of computer-aided molecular design, 8(3), 243-256. https://doi.org/10.1007/BF00126743, PMid:7964925
Acharya, C., Coop, A., E Polli, J., & D MacKerell, A. (2011). Recent advances in ligand-based drug design: relevance and utility of the conformationally sampled pharmacophore approach. Current computer-aided drug design, 7(1), 10-22. https://doi.org/10.2174/157340911793743547 , PMid:20807187 , PMCid:PMC2975775
Lin, J. H. (2016). Review structure‐and dynamics‐based computational design of anticancer drugs. Biopolymers, 105(1), 2-9. https://doi.org/10.1002/bip.22744, PMid:26385494
Prada-Gracia, D., Huerta-Yépez, S., & Moreno-Vargas, L. M. (2016). Application of computational methods for anticancer drug discovery, design, and optimization. Boletín Médico Del Hospital Infantil de México (English Edition), 73(6), 411-423. https://doi.org/10.1016/j.bmhime.2017.11.040, https://doi.org/10.1016/j.bmhimx.2016.10.006, PMid:29421286
Dev Bukhsh Singh (2014) Success, Limitation and Future of Computer Aided Drug Designing. Transl Med (Sunnyvale) 4: E-127. doi:10.4172/2161-1025.1000e127
Durrant, J. D., & McCammon, J. A. (2011). Molecular dynamics simulations and drug discovery. BMC biology, 9(1), 71. https://doi.org/10.1186/1741-7007-9-71 , PMid:22035460, PMCid:PMC3203851
Read the full article
0 notes
Text
Formulation and Evaluation of Fast Dissolving Tablet of Lamotrigine
INTRODUCTION
Oral routes of drug administration have wide acceptance up to 50-60% of total dosage forms. Solid dosage forms are popular because of ease of administration, accurate dosage, self-medication, pain avoidance and most importantly the patient compliance. The most popular solid dosage forms are being tablets and capsules; one important drawback of this dosage forms for some patients, is the difficulty to swallow. Drinking water plays an important role in the swallowing of oral dosage forms.
Often times people experience inconvenience in swallowing conventional dosage forms such as tablet when water is not available, in the case of the motion sickness (ketosis) and sudden episodes of coughing during the common cold, allergic condition and bronchitis. For these reason, tablets that can rapidly dissolve or disintegrate in the oral cavity have attracted a great deal of attention. Or dispersible tablets are not only indicated for people who have swallowing difficulties, but also are ideal for active people4. Fast dissolving tablets areal so called as mouth-dissolving tablets, melt-in mouth tablets, Orodispersible tablets, rapid melts, porous tablets, quick dissolving etc. Fast dissolving tablets are those when put on tongue disintegrate instantaneously releasing the drug which dissolve or disperses in the saliva5. The faster the drug into solution, quicker the absorption and onset of clinical effect. Some drugs are absorbed from the mouth, pharynx and oesophagus as the saliva passes down into the stomach. In such cases, bioavailability of drug is significantly greater than those observed from conventional tablets dosage form. The advantage of mouth dissolving dosage forms are increasingly being recognized in both, industry and academics7. Their growing importance was underlined recently when European pharmacopoeia adopted the term ―Orodispersible tablet‖ as a tablet that to be placed in the mouth where it disperses rapidly before swallowing. According to European pharmacopoeia, the ODT should disperse/disintegrate in less than three minutes. The basic approach in development of FDT is the use of superdisintegrants like cross linked carboxy methyl cellulose (crosscarmellose), sodium starch glycolate (primogel, explotab), polyvinyl pyrollidon (polyplasdone) etc, which provide instantaneous disintegration of tablet after putting on tongue, their by release the drug in saliva. The bioavailability of some drugs may be increased due to absorption of drug in oral cavity and also due to pre gastric absorption of saliva
Containing dispersed drugs that pass down into the stomach. More ever, the amount of drug that is subject is to first pass metabolism is reduced as compared to standard tablet. The technologies used form manufacturing fast-dissolving tablets are tablet sublimation.
Following conventional techniques are used for preparation of fast dissolving drug delivery system7-9
Sublimation
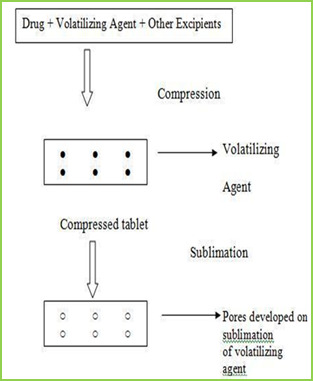
The slow dissolution of the compressed tablet containing even highly water-soluble ingredients is due to the low porosity of the tablets. Inert solid ingredients that volatilize readily (e.g. urea, ammonium carbonate, ammonium bicarbonate, hexa Methylene tetramine, camphor etc.) were added to the other tablet ingredients and the mixture is compressed into tablets. The volatile materials were then removed via sublimation, which generates porous structures. Additionally, several solvents (e.g. cyclohexane, benzene) can be also used as pore forming agents,
MATERIAL & METHODS
Table no-1
Sr no.
Name of Ingredient
Supplier
1
Lamotrigine
Abottpharmapvt. Ltd., Goa
2
Sodium StarchGlycollate
Yash Scientific Enterprises, Pune
3
Crosscarmellose Sodium
Kurla Complex, Mumbai
4
β-Cyclodextrin
Ozone international, Mumbai
5
Aerosil
Yash Scientific Enterprises, Pune
6
Camphor
Yash Scientific Enterprises, Pune
7
Directly Compressible Lactose
Yash Scientific Enterprises, Pune
Method
Preformulation Study
Organoleptic Characteristics
Physico-chemical Characterization
1 Bulk Density
2 Tapped Density
3 Carr‘s index
4 Hausner‘s Ratio
5 Angle of Repose
Calibration curve of Drug
Formulation & Evaluation of Tablet
Hardness
Disintegration Time
Thickness
Friability
Wetting Time
Drug Content
Weight Variation
8. Invitro Drug Release (Dissolution Study)
Formulation procedure of tablet (direct compression)
In process of direct compression techniques, the all ingredients were accurately weighed and passed through sieve no.40 then mixed together and then compressed using 6 mm flat punch on Cemach R&D Tablet press 10 station compression machine. Hardness of the tablet was maintained at 3-3.5 Kg/cm2. Tablet weight was maintained at 170 to 180 mg. All the product and process variables like mixing time and hardness were kept as practically constant.
Table no-2
Sr No.
Name of
ingredients
F1 (mg)
F2 (mg)
F3 (mg)
F4 (mg)
F5 (mg)
F6 (mg)
1
Lamotrigine
25
25
25
25
25
25
2
β-cyclodextrin
25
25
25
25
25
25
3
Sodium starch
21
26.25
31.5
-
-
-
glycolate
4
Crosscarmellose
-
-
-
21
26.25
31.5
sodium
5
Direct
compressible
10.7
5.45
0.2
10.7
5.45
0.2
6
Camphor
7
7
7
7
7
7
8
Total weight
90
90
90
90
90
90
RESULTS AND DISCUSSION
In this study fast dissolving tablet of Lamotrigine were prepared by direct compression. Method and effect of different superdisintegrating and sublimating agent camphor on in vitro release were evaluated.
Organoleptic Characteristics
Organoleptic characteristics like colour, odour, and taste were studied. The Lamotrigine complies with specifications. The results are illustrated in table
Table No.3 Organoleptic properties of Lamotrigine
Sr. No
Properties
Specification
Lamotrigine
1
Appearance
White
White
2
Description
Crystalline
Crystalline
3
Odour
Odourless
Odourless
4
Taste
Bitter
Bitter
Physical characterization
The powder bed was evaluated for the blend property like Bulk density, Tapped density, Carr‘s index, Hausner‘s ratio and Angle of repose.
Batch
code
Bulk density
(gm/ml) ± SD
Tapped density
(gm/ml) ± SD
Carr’s index
% ± SD
Hausner’s ratio
% ± SD
Angle of
repose (0) ± SD
F1
0.6032±0.03
0.6912 ± 0.01
14.25 ± 0.20
1.1124 ± 0.02
20.07 ± 0.54
F2
0.6133 ± 0.05
0.6999 ± 0.02
14.09 ± 0.39
1.1358 ± 0.07
19.45 ± 0.85
F3
0.6258 ± 0.01
0.7134 ± 0.06
15.00 ± 0.13
1.1425 ± 0.06
19.39 ± 0.29
F4
0.6078 ± 0.07
0.7088 ± 0.09
15.04 ± 0.75
1.1298 ± 0.04
20.14 ± 0.17
F5
0.6125 ± 0.02
0.7032 ± 0.05
14.58 ± 0.09
1.1340 ± 0.03
20.73 ± 0.65
F6
0.6289 ± 0.08
0.7155 ± 0.04
14.99 ± 0.67
1.1536 ± 0.01
20.10 ± 0.44
Calibration curve of Drug
Stock solution of 100 µg/ml was prepared in 0.1 ml N HCL, from which dilution were made to obtain 2, 4, 6, 8, 10 µg/ml solution. Absorbance of these solutions when measured at ƛmax 267 nm and the results are given Table.
Table No 5 Calibration curve of lamotrigine in 0.1 N HCL
Sr. No.
Concentration (µg/ml)
Absorbance at 267 nm ±SD
1
0
0 ± 00
2
2
0.1927± 0.00015
3
4
0.2360± 0.00023
4
6
0.2924± 0.00011
5
8
0.3207± 0.00046
6
10
0.3913± 0.00078
Calibration curve
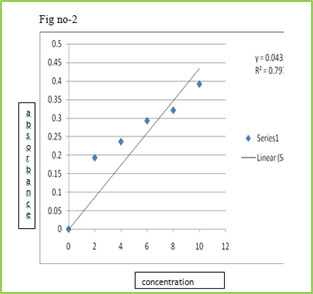
Figure 2: Standard calibration curve of Lamotrigine in 0.1 N HCl
Evaluation of compression characteristics of formulations
Tablets of all batches were evaluated for weight variation, hardness, thickness and friability results were tabulated in Table.
Tablet No.6 Post compression properties of tablets F1 to F6
Batch
code
Weight variation
(mg) ± SD
Hardness (kg/cm2)
± SD
Thickness (mm)
± SD
Friability ±
SD%
F1
0.090 ± 0.02
12.25 ± 0.28
5.02 ± 0.03
0.63 ± 0.02
F2
0.090 ± 0.01
13.20 ± 0.90
5.05 ± 0.08
0.52 ± 0.02
F3
0.090 ± 0.03
13.00 ± 0.26
5.06 ± 0.02
0.73 ± 0.01
F4
0.090 ± 0.02
13.10 ± 0.13
5.07 ± 0.05
0.89 ± 0.03
F5
0.090 ± 0.03
13.23 ± 0.58
5.03 ± 0.04
0.75 ± 0.04
F6
0.090 ± 0.01
13.05 ± 0.10
5.04 ± 0.01
0.45 ± 0.03
Evaluation of various Parameters of Tablets
The tablets were evaluated for disintegration time, wetting time, and drug content. Results obtained were given in Tablet.
Table No.7 other post compression parameters of tablets F1 to F6
Batch
code
Disintegration
time (s)± SD
Wetting Time (s)
± SD
Drug content
± SD
F1
58.05 ± 0.07
62.37 ± 0.54
95.49 ± 1.11
F2
53.14 ± 0.04
59.48 ± 0.34
96.76 ± 0.92
F3
45.38 ± 0.03
56.35 ± 0.12
98.48 ± 1.07
F4
15.20 ± 0.10
57.01 ± 0.89
97.68 ± 1.15
F5
17.02 ± 0.07
55.42 ± 0.45
95.21 ± 1.01
F6
08.20 ± 0.03
53.32 ± 0.75
101.23 ± 1.05

Figure 3: wetting time of fast dissolving tablet of Lamotriginre (Tablet wetting initial, Tablet wetting after 53.32 sec)
In vitro Drug Release (Dissolution Study)
Dissolution test were carried out using USP Type dissolution test apparatus at 37 ± 0.50C and rpm speed. 900 ml of 0.1 N HCl was used as dissolution medium. Two tablets from each tablets were tested individually in 0.1 N HCl with sample withdraw 5 ml. Collected samples were analysed at ƛ max 267 nm using 0.1 N HCl as blank. The percentage drug release was found to formulation F1 to F6 are given in following tables.
Table No.7 In vitro drug release data of formulation F1 (n=2)
Time
(min)
Absorbance
(267nm)
Concentration
(µg/ml)
Cumulative
drug release
Percentage
CDR (%)
0
0
0
0
0
1
0.0415
1.22
0.048
4.8
2
0.1737
5.18
0.20
20.43
5
0.3995
11.75
0.47
47.00
15
0.5805
17.07
0.68
68.28
30
0.8298
24.40
0.97
97.60
Table No.8 In vitro drug release data of formulation F2 (n=2)
Time
(min)
Absorbance
(267nm)
Concentration
(µg/ml)
Cumulative
drug release
Percentage
CDR (%)
0
0
0
0
0
1
0.0761
2.23
0.08
8.9
2
0.1908
5.61
0.22
22.44
5
0.38.37
11.28
0.45
45.12
15
0.5638
16.58
0.66
66.32
30
0.8344
24.54
0.98
98.16
Table No.9 In vitro drug release data of formulation F3 (n=2)
Time
(min)
Absorb-ance
(267nm)
Concn
(µg/ml)
Cumulative
drug release
Percentage
CDR (%)
0
0
0
0
0
1
0.0625
1.83
0.07
7.35
2
0.2248
6.61
0.26
26.44
5
0.4158
12.22
0.48
48.88
15
0.5960
17.52
0.70
70.08
30
0.8495
24.98
0.99
99.92
Table No.10 In vitro drug release data of formulation F4 (n=2)
Time
(min)
Absor-bance
(267nm)
Concn
(µg/ml)
Cumulative
drug release
Percentage
CDR (%)
0
0
0
0
0
1
0.0305
0.89
0.03
3.5
2
0.2348
6.90
0.27
27.62
5
0.4009
11.79
0.47
47.16
15
0.6010
17.67
0.70
70.68
30
0.8489
24.96
0.99
99.84
Table No.11 in vitro drug release data of formulation F5 (n=2)
Time
(min)
Absorbance
(267nm)
Concen
(µg/ml)
Cumulative
drug release
Percentage
CDR (%)
0
0
0
0
0
1
0.0238
0.7
0.02
2.8
2
0.1983
5.83
0.23
23.32
5
0.3785
11.13
0.44
44.52
15
0.5969
17.55
0.70
70.20
30
0.8239
24.23
0.96
96.92
Table No.12 In vitro drug release data of formulation F6 (n=2)
Time
(min)
Absorb-ance
(267nm)
Concn
(µg/ml)
Cumulative
drug release
Percentage
CDR (%)
0
0
0
0
0
1
0.0843
2.47
0.09
9.9
2
0.2248
6.61
0.26
26.44
5
0.4475
13.16
0.52
52.64
15
0.6308
18.55
0.74
74.20
30
0.8399
24.70
0.98
98.81
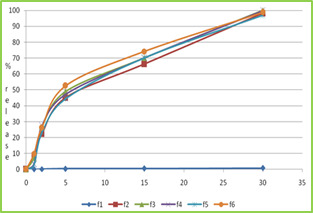
Figure no-4 dissolution profile of formulation F1 to F6
CONCLUSION
The results obtained so far encouraged as to derive following conclusion,
Fast dissolving tablet of Lamotrigine was formulated by using various superdisintegrants like Crosscarmellose sodium and Sodium starch glycolate in different proportions by sublimating agent like camphor.
The values of pre-compression parameters of all formulation showed good flow properties and compressibility, so these can be used for tablet manufacture.
The disintegration time for all formulations was considered to be within the acceptable limit. It observed that when sublimating agent like camphor was used disintegration time of tablet is decreased.
Wetting time studies showed that wetting time was rapid in formulations containing camphor followed by CCS and SSG. It was found that as the concentration of CCS and SSG was increases, then wetting was reduces.
The post compression parameters of all formulations were determined and the values were found to be within IP limits.
In-vitro disintegration of F3 gives rapid disintegrating time and wetting time.
As result of this study, it may be concluded inclusion the complexation techniques may be useful to enhance solubility and dissolution rate.
The concept of formulating high porous fast dissolving tablets of Lamotrigine inclusion complexes using superdisintegrants by sublimation technique offers a suitable and practical approach in serving desired objectives of faster disintegration and dissolution characteristics.
REFERENCES
Lachman L, Lieberman HA, Kanig JL. The theory and practice of industrial pharmacy.
Third Edition, Varghese Publication House, Bombay, India 1987: 296‐
Amin, A. F., Shah, T. J., Bhadani, M. N., & Patel, M. M. (2005). Emerging trends in orally disintegrating tablets.
Prakash Goudanavar et al. (2011). Development and characterization of lamotrigineorodispersible tablets Inclusion complex with hydroxypropyl β cyclodextrin‖ International Journal of Pharmacy and Pharmaceutical Sciences, 3(3), 208-214
Seager, H. (1998). Drug‐delivery products and the Zydis fast‐dissolving dosage form. Journal of pharmacy and pharmacology, 50(4), 375-382. https://doi.org/10.1111/j.2042-7158.1998.tb06876.x, PMid:9625481
Remon, J. P., & Corveleyn, S. (2000). S. Patent No. 6,010,719. Washington, DC: U.S. Patent and Trademark Office.
Masaki, K., Intrabuccaly disintegrating preparation and production thereof, US Patent No.5, 466, 464, 1995.
Pebley, W.S., Jager, N.E., Thompson, S.J., Rapidly disintegrating tablets, US Patent No.5, 298, 261, 1994.
Amrutkar, P. P., Patil, S. B., Todarwal, A. N., Wagh, M. A., Kothawade, P. D., & Surawase, R. K. (2010). Design and evaluation of taste masked chewable dispersible tablet of lamotrigine by melt granulation. International Journal of Drug Delivery, 2(2). https://doi.org/10.5138/ijdd.2010.0975.0215.02028
Allen Jr, L. V., & Wang, B. (1996). S. Patent No. 5,587,180. Washington, DC: U.S. Patent and Trademark Office.
Biradar, S. S., Bhagavati, S. T., & Kuppasad, I. J. (2006). Fast dissolving drug delivery systems: a brief overview. The internet journal of pharmacology, 4(2), 26-30.
Zade, P. S., Kawtikwar, P. S., & Sakarkar, D. M. (2009). Formulation, evaluation and optimization of fast dissolving tablet containing tizanidine hydrochloride. Int J Pharm Tech Res, 1(1), 34-42.
Sukhavasi, S., & Kishore, V. S. (2012). Formulation and evaluation of fast dissolving tablets of amlodipine besylate by using hibiscus rosa-sinensis mucilage and modified gum karaya. International Journal of Pharmaceutical Sciences and Research, 3(10), 3975.
Patil, C., & Das, S. (2009). Effect of various superdisintegrants on the drug release profile and disintegration time of Lamotrigine orally disintegrating tablets. African journal of pharmacy and pharmacology, 5(1), 76-82. https://doi.org/10.5897/AJPP10.279
Swamy, P. V., Areefulla, S. H., Shirs, S. B., Smitha, G., & Prashanth, B. (2007). Orodispersible tablets of meloxicam using disintegrant blends for improved efficacy. Indian journal of pharmaceutical sciences, 69(6), 836. https://doi.org/10.4103/0250-474X.39448
Shah S. D. (2010). M.Pharm thesis, Formulation and evaluation ofsublingual tablet of Sumatriptan succinate, Saurashtra University.
Read the full article
0 notes
Text
Analytical Method Development and Validation for Sultamicillin Tosylate Dihydrate in Bulk and Pharmaceutical Dosage Forms by RP-HPLC
INTRODUCTION
Sultamicillin Tosylate Dihydrate, chemically known as (2S,5R)-(3,3-Dimethyl-4,4,7-trioxo-4-thia-1-azobicyclohept-2-ylcarbonyl)methyl(2S,5R,6R)-6--3,3dimethyl-7-ox0-4-thia-1-azabicycloheptanes-2-carboxylatemono-4-tolunesulfonate dihydrate. This is a mutual (joint) prodrug of Ampicillin and Sulbactam compounds attached together with ester connection.
Fig.1: Chemical structure of Sultamicillin Tosylate Dihydrate
This mutual prodrug is one of the antibiotics with plenty antimicrobial spectrum for the treatment of childhood pneumonia. The irretrievable β-lactamase inhibitor sulbactam has been combined chemically via ester linkages with ampicillin to form sultamicillin. It was composed of double esters of formaldehyde hydrate in which one of the hydroxyl groups is esterified with ampicillin and sulbactam. It is hydrolyzed quickly in neutral or faintly alkaline conditions, while hydrolyzed; it forms ampicillin and hydroxylmethyl sulbactam or sulbactam and hydroxylmethyl ampicillin by different routes.It is available obtainable in both oral and parenteral preparations for child (pediatric) use. Sultamicillin is also a valuable treatment option for a multiplicity of paediatric infections, bacterial infections in children including those due to β-lactamase-producing organisms. The use of β-lactam and β-lactamase inhibitor mixtures, particularly ampicillin and sulbactam, as empiric treatment or prophylaxis for number of pediatric infections are healthy established, and have been extensively reviewed over number of years. The antimicrobial action of Sultamicillin had been established in vitro against extensive range of gram-positive and negative organisms and as well as anaerobes .
Sultamicillin Tosylate Dihydrate is a white crystalline powder which is freely soluble in methanol, acetonitrile, acetone and insoluble in water, benzene, chloroform, diethyl ether.
The present work is to develop and validate RP-HPLC method for the determination of Sultamicillin Tosylate Dihydrate in API and its Pharmaceutical Dosage Form.
MATERIAL & METHODS
A calibrated weighing balance (Shimadzu) of 1 mg sensitivity was used.
A HPLC Younglin Acme 9000 series quaternary gradient pump SP 930D. HPLC system accomplished with UV 370D UV Visible detector with 20µl Rheodyne injector. Data was processed on Autochrome-3000 software. Column C18 (150 x 4.6 mm, 5µm) phenomenex with UV method analysis was performed on UV visible double beam spectrophotometer Shimadzu 1800.
Mobile phase filtered through a Nylon 6,6 membrane 0.45 µm, 47mm filters (pall India Pvt.Ltd. Mumbai) using vacuum pump. Ultra sonicator (Microlean-103) was used for degassing the mobile phase.
The solutions were filtered through 0.45 µ syringe filter (Phenomenex).
Chemicals
Sultamicillin Tosylate Dihydrate drug powder was gifted by Associated Biotech, Vill. Kishanpura, Gurumajra Road, Baddi, India. Sultamicillin Tosylate Dihydrate tablets of 375 mg strength were purchased from the local pharmacy in Solapur under commercially available brand name Marzon (Eris Lifesciences Limited).
Acetonitrile LiChrosolv®, water LiChrosolv® was purchased from Merck Specialities Pvt. Ltd, Mumbai.
Method
Chromatographic Conditions
The chromatographic separation was performed by analytical column: phenomenex C18 column (150 x 4.6 mm, 5µm) using mobile phase acetonitrile:water(45:55) at flow rate 1.0 ml/min. with isocratic elution. The injection volume was 20 µl and the run time was 10 minute. Detection was carried out at 225 nm.
Preparation of standard stock solution:
The standard stock solution of Sultamicillin Tosylate Dihydrate was prepared by transferring, accurately weighed 10 mg of Sultamicillin Tosylate Dihydrate to 10 ml of volumetric flask containing 5ml methanol and dissolved. Then volume was made up to the mark by using methanol to gives concentration 1000 µg/ml. From this 1ml of the solution was transferred to a 10 ml volumetric flask and make up the volume with mobile phase (ACN:Water) to get a concentration of 100 µg/ml of sultamicillin Tosylate Dihydrate and labelled as “Standard stock solution”.
Tablet solution of Sultamicillin Tosylate Dihydrate
Tablet powder weight containing equivalent 10 mg of sultamicillin Tosylate was weighed and transferred to a 10 ml volumetric flask then dissolved in the methanol LR. The volume was made up to the mark with same solvent to obtain conc. of 1000µg/ml of Sultamicillin Tosylate Dihydrate. From the resulting solution 1 ml was diluted to 10 ml with the ACN:Water (4.5:5.5) solvent to obtain conc. of 100µg/ml of Sultamicillin Tosylate Dihydrate , and labeled as ‘Std Stock Tablet Sultamicillin Tosylate Dihydrate’.
Assay of sultamicillin Tosylate Dihydrate Tablet
20 tablets weighed and powdered. The powder equivalent to 10 mg of sultamicillin Tosylate dihydrate was weighed transferred into100 ml volumetric flask and dissolved in methanol LR. Solution was sonicated for 15 minutes and final volume was made up to the mark with methanol LR. 1ml of solution was transferred into 10 ml of volumetric flask and diluted up to 10ml with mobile phase and sample was analysed.
Selection of wavelength
The standard solution of 100µg/ml was scanned in the UV range 200-400nm. The solution showed maximum absorption at 225nm.
Validation of RP-HPLC Method
Specificity
The chromatogram of standard solution of Sultamicillin Tosylate Dihydrate was compared with chromatogram of its degradants.
Linearity
From the ‘Std Stock Sultamicillin Tosylate Dihydrate’ (100µg/ml) solution, the volume quantity of 1, 2, 4, 5 and 6 ml were transferred in a series of 10ml volumetric flasks. The volume was made up to the mark with mobile phase to obtain the concentration of 10, 20, 40, 50 and 60µg/ml of Sultamicillin Tosylate Dihydrate .
The solutions were filtered through syringe filter and 20µl injected into the HPLC system and their chromatogram were recorded for 10mins. Under the chromatographic conditions as described above after getting a stable baseline. Peak areas were recorded for all the peaks. Calibration curve of Sultamicillin Tosylate Dihydrate was constructed by plotting the peak area of Sultamicillin Tosylate Dihydrate v/s conc. of Sultamicillin Tosylate Dihydrate. The correlation coefficient (r2) of least square linear regression for Sultamicillin Tosylate Dihydrate was calculated.
III. Range
The range of analytical method was decided from the interval between upper and lower level of calibration curves by plotting the curve. The correlation coefficient (r2) of least square linear regression for Sultamicillin Tosylate Dihydrate was calculated.
Precision
The precision of an analytical method was studied by performing Repeatability and intermediate precision.
a) Repeatability: From the ‘Std Stock Sultamicillin Tosylate Dihydrate’ (100µg/ml) solution, 2ml was transferred in 10ml volumetric flasks. The volume was made up to the mark with mobile phase to obtain the conc. of 20µg/ml of Sultamicillin Tosylate Dihydrate. The solution was filtered through syringe filter and 20µl injected into the HPLC system and its chromatogram was recorded under the same chromatographic conditions after getting a stable baseline. Peak area was recorded. The procedure was repeated for thrice.
Limit of Detection
Detection limit was determined based on the standard deviation of peak areas of same concentrations i.e. Standard solution of Sultamicillin Tosylate Dihydrate (20µglml) prepared six times and LOD calculated by the following formulae.
LOD = 3.3(SD/S)
Where, SD- Standard deviation; S- Slope of Curve
Limit of Quantitation
Quantitation limit was determined based on the standard deviation of peak areas of same concentrations i.e. Standard solution of Sultamicillin Tosylate Dihydrate (20µglml) prepared six times and LOQ calculated by the following formulae.
LOQ calculated by the following formulae.
LOQ = 10(SD/S)
Where, SD- Standard deviation; S- Slope of Curve
VII. Robustness
The standard solution of (20µg/ml) was prepared and analyzed at different flow rates (0.9, 1.0, 1.1 ml/min) and at different wavelengths (224, 225, and 226).
VIII. System Suitability
Sample solutions of Sultamicillin Tosylate Dihydrate (50µg/ml) were prepared and analyzed six times. Chromatograms were studied for different parameters such as tailing factor, resolution and theoretical plates to see that whether they comply with the recommended limit or not.
Accuracy
Recovery study was carried out by standard addition method by adding the known amount of sultamicillin Tosylate dehydrate to preanalysed sample at three different conc. level i.e. 80%, 100%, 120% of assay conc. and percent recovery were calculated.
0.5 ml tablet solution was transferred to 4 different 10ml volumetric flasks (Labelled as blank, 80%, 100%, 120%) separately and 0, 1.6, 2, 2.4ml of 100µg/ml ‘standard solution’ was added respectively and the volume was made up to the mark with mobile phase. and these samples were analysed.
RESULTS AND DISCUSSION
Determination of wavelength of maximum absorption
The wavelength of maximum absorption was found to be 225 nm. Hence HPLC analysis was carried out at 225nm.
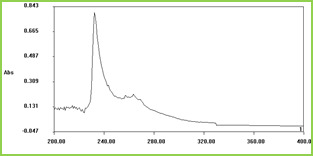
Fig 2: Wavelength of maximum absorption of Sultamiciilin Tosylate
Specificity

Fig 3: Chromatogram of Sultamicillin Tosylate Dihydrate with degradants
Linearity
The linearity of this method was determined at the range from 10-60µg/ml for Sultamicillin Tosylate Dihydrate.
The regression equation was found to be Y=8.7304x+5.5409 be, r2=0.9991.
Table No-1: Linearity table
Sr. No
Concentration (µg/ml)
Peak Area(mV)
1
10
98.48
2
20
176.48
4
40
349.73
4
50
438.66
5
60
535.84
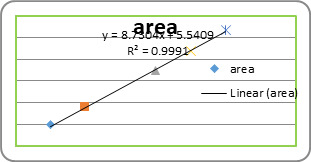
Fig. 3: Linearity graph of Sultamicillin Tosylate Dihydrate

Fig.4: 0verlain chromatograms of serial dilutions of Sultamicillin Tosylate Dihydrate in optimized chromatographic conditions
The linearity for Sultamicillin Tosylate Dihydrate was found to be linear in the range of 10-60µg/ml with r2= 0.9991 and the straight line equation as Y= 8.7304x+5.5409.
4.4 Range:
The range for RP-HPLC method for Sultamicillin Tosylate Dihydrate was found to be 10-60 µg/ml.
4.5 Precision
The precision was evaluated as the repeatability of the method and calculated as %RSD values for six determinations of peak area ratio performed on the same day and under the same experimental condition.
4.5.1 Repeatability
Table No-2: Repeatability study for Sultamicillin Tosylate Dihydrate
Injection
Peak Area of sultamicllin Tosylate Dihydrate (mV)
1
176.47
2
172.61
3
178.99
4
176.28
5
173.81
6
174.41
SD
2.284508
%RSD
1.30
The percentage RSD (˂2) Values obtained shows that the method developed in précised at repeatability.
4.6 Limit of Detection
Detection limit is calculated based on standard deviation of response and slope
Table No-3: Limit of Detection Data of Sultamicillin Tosylate Dihydrate
Sultamicillin Tosylate Dihydrate
LOD(µg/ml)
0.02754
4.7 Limit of Quantification
Quantification limit is calculated based on standard deviation of response and slope
Table No-4: Limit of Quantification data of Sultamicillin Tosylate Dihydrate
Sultamicillin Tosylate Dihydrate
LOD(µg/ml)
0.08345
4.8 Robustness
The robustness was investigated by achieving deliberate changes in flow rate by ±1 units from 1.1 to 0.9ml/min and change in wavelength by ±1nm that flow is at 225nm Robustness of the method was carried out at concentration of 20µg/ml and then T, Rs and N were evaluated. The system suitability parameters remained unaffected over deliberate small change in the chromatoghraphic conditions, illustrating that the method was robust over an acceptable working range of its HPLC operational parameters.
Table No-5: Result of Robustness Study: Variation in flow rate and wavelength
Sr no.
Conditions
Range
Investigated
Retention
Time (min)
Theorotical
Plates(N)
Resolution
Tailing Factor(T)
1
Flow Rate
(ml/min)
1.1
6.15
7129.9
3.56
1.22
0.9
7.58
8195.1
8.36
1.26
2
Wavelength
(nm)
226
6.88
9493.3
7.91
1.25
224
6.85
5013.8
2.138
1.08
System Suitability Testing
Study of resolution, tailing factor and capacity factor shows system is suitable for this method
Table No-5: Results of System Suitability Parameters
Analyte
Retention Time (min)
Tailing Factor (T)
Theoretical
Plates (N)
Resolution
(R)
Sultamicillin Tosylate Dihydrate
6.9
1.34
7349.6
7.64
Required limits
--
T N > 2000
R >2
Assay of Sultamicillin Tosylate Dihydrate
Table No-6: Results of Assay
Tablet Formulation
Lable claim
Amount taken
µg
Amount found
µg
Assay%
Marzon
375 mg
37
38
102%
Accuracy
The accuracy study of method was determined through the recovery test of the samples, using known amounts of sultamicillin Tosylate dehydrate reference standard.
Table No-7: Results of accuracy of Sultamicillin Tosylate Dihydrate
Sr.no
Level of % Recovery
Amount of Tablet sample solution
(ml)
Amount of standard drug added (µg/ml)
Amount Added
µg
Amount
Found
(µg/ml)
% Recovery
1
0
0.5
0
0
0
-
2
80
0.5
1.6
16
16.28
101
3
100
0.5
2
20
20.74
103
4
120
0.5
2.4
24
24.75
103
SUMMARY AND CONCLUSION
Summary
Analytical method development was started with preliminary studies of the Sultamicillin Tosylate Dihydrate according to BP. The drug is freely soluble in methanol. The stock solutions of the drug were prepared in methanol.
The RP-HPLC method for estimation of Sultamicillin Tosylate Dihydrate dosage form was developed. The quantification was carried out by using Phenomenex C18 column (150 mm × 4.6 mm, 5 mm) as stationary phase and acetonitrile: water (45:55) as mobile phase. Mobile phase was maintained at a flow rate of 1.0ml/min. The UV detector was operated at 225 nm and Sultamicillin Tosylate Dihydrate eluted at 6.90 min.
The developed RP-HPLC method can be successfully applied for the routine analysis of Sultamicillin Tosylate Dihydrate.
Table No-8: Summary of RP-HPLC Method of Sultamicillin Tosylate Dihydrate
Sr. No.
Parameters
Sultamicillin Tosylate Dihydrate
1.
Linearity Range (μg/ml)
10-60
2.
Regression Equation
(y = mx+c)
8.730x+5.540
3.
Correlation Coefficient (r2)
0.999
4.
LOD (μg/ml)
0.02754
5.
LOQ (μg/ml)
0.08345
6.
Repeatability (%RSD)
1.06
7.
Robustness(%RSD)
Flow Rate
Wavelength
11.10%
23.94%
Conclusion
In conclusion, the proposed HPLC method is simple, accurate, reproducible method for estimation of Sultamicillin Tosylate Dihydrate in bulk and pharmaceutical formulation. This method shows Assay of Sultamicillin Tosylate Dihydrate within the specified limit. The method shows no interference by the excipients. The statistical parameters and recovery data reveals the good accuracy and precision of the proposed method. Finally, since no pharmacopoeial method for determination of Sultamicillin Tosylate Dihydrate in bulk and pharmaceutical formulations have been reported yet, the proposed method could be useful and suitable for the estimation of the Sultamicillin Tosylate Dihydrate in bulk and pharmaceutical dosage forms.
ACKNOWLEDGEMENT:
The authors are grateful to Principal of D.S.T.S. Mandal’s College of pharmacy, Solapur, Maharashtra, India for providing us the research facility. Drug powder was gifted by Associated Biotech and grateful to Mr. Vichitra (Medicef Pharma)
REFERENCES
Nahler, G. (2009). International Conference on Harmonisation (ICH). In Dictionary of Pharmaceutical Medicine(pp. 96-96). Springer, Vienna. https://doi.org/10.1007/978-3-211-89836-9_719
Sadhana, k., Vasanth, P. M, Ramesh, T, Ramesh Malothu. (2013). Development of New Validated Method for the Determination OF Sultamicillin Tosylate Dihydrate in Tablet Dosage Forms By RP-HPLC. IJPSR, 3(1), 14-16.
Kumar, V. J., Gupta, P. B., Kumar, K. P., Ray, U. K., Sreenivasulu, B., Kumar, G. S., & Mukkanti, K. (2011). Identification, isolation and characterization of a new degradation product in sultamicillin drug substance. Journal of pharmaceutical and biomedical analysis, 54(3), 582-587. https://doi.org/10.1016/j.jpba.2010.09.016, PMid:20934824
Japanese Pharmacoepoeia 15 Edition. 1- 1654.
https;//en.wikipedia.org/sultamicillin.
S. Muvvala, V.N. Rantakaram and R.R. Nadendla. (2012) A Validated RP-HPLC Method for the Estimation of Febuxostat in Bulk Drugs. Int. J. Pharm tech. Res., 4(4), 1352-1366.
Lasan V. M, Patel S. A. (2015). Analytical Method Development And Validation for Escitalopran oxalate in pharmaceutical Dosage forms by HPLC Methods. International Journal for Pharmaceutical Research Scholars, 4(1), 19-24.
Dinesh Reddy Salla, Vindhyakini dhanthala. (2017). A Validated RP-HPLC Method for Estimation of Rifaximin in its Bulk and Pharmaceutical Dosage forms. International Journal for Pharmaceutical Research Scholars, 6(1), 12-20.
S. Potdar, M. S. Kalshetti, R.Y. Patil. (2017). Development and Validation of Novel RP-HPLC for Simultaneous Estimation of Alogliptin Benzoate and Pioglitazone Hcl in Pharmaceutical dosage form. International Journal of Chemical and Pharmaceutical Analysis, 4(3), 1-9.
Read the full article
0 notes
Text
Formulation and Characterization of Rivastigmine Loaded Solid Lipid Nanoparticles
INTRODUCTION
Alzheimer disease (AD) is most common prevalent neurodegenerative disorder. Today, it affects nearly 30 million people in the whole world. With each passing year about 4 million people in the world develop dementia. As the average population increases, the number of AD patients is expected to rise exponentially and about 110 million of patients are projected for 2050.
There are some common features suggesting that in AD brain could be an acceleration of processes occurring in aged brain. Adult neurogenesis occurring in the dentate gyrus (DG), a process that decreases in aged mammals and that could be related with loss of memory, an important feature in AD.
A loss in declarative memory has been found in patients with AD. In these patients, neurodegenerative at the hippocampal region takes place at the first steps of the disease. In normal ageing there is a mild cognitive impairment, but this impairment could be accelerated in AD.1
Alzheimer’s disease (AD) applied to a state of presenile dementia, extra-neuronal protein aggregations (plaques), and intraneuronal protein aggregations (tangles). Although it was recognized at the time that brains of persons with senile dementia could also manifest plaques and tangles, in the elderly this was not felt to represent an actual disease state.2
Alzheimer’s disease (AD) is a chronic and progressive neurodegenerative disorder that begins with cognitive and memory impairments, accompanied with behavioural disturbances such as aggression, depression, hallucination, delusion, anger and agitation and eventually progresses to dementia, physical impairment and death.3
Rivastigmine Tartrate
Chemically Rivastigmine tartrate is N-Ethyl-N-methylcarbamic acid 3-phenyl ester (2R,3R)-2,3-dihydroxybutanedioate.4
Figure 1: Structure of Rivastigmine Tartrate
Rivastigmine tartrate is a white to off-white powder.5 It is very soluble in water, soluble in ethanol and acetonitrile, slightly soluble in n-octanol and very slightly soluble in ethyl acetate. It has molecular formula C14H22N2O22.C4H6O6 having molecular weight 400.43 g/mol.6
Rivastigmine tartrate is a reversible (or pseudoirreversible because it separates too slowly from AChE) nonselective cholinesterase inhibitor which inhibits both AChE and BuChE in the central nervous system (CNS). It binds both esteratic and ionic sites of AChE just like a natural substrate, and it inhibits the metabolism of Ach. It is 4-6 times more effective on the G1 (monomeric) form of the enzyme, which is present at higher concentrations in the brain of AD patients. There is no affinity of rivastigmine tartrate for muscarinic, alpha- or beta-adrenergic, or dopamine receptors or opoid binding sites.7
Aim and objective of present research work
Presently rivastigmine tartrate is available in the form of tablet, capsule containing 1.5mg, 3mg, 4.5mg, 6mg and the common side effects associated with oral administration (gastrointestinal) like vomiting, diarrhoea, increased acid secretion in stomach and reduced heart rates. Oral administration shows significant first-pass effect. Its half-life is about 1.5 hrs.
Rivastigmine tartrate is also available in the form of transdermal patch containing 4.5mg, 9.5mg and the common side effects associated with transdermal route are allergic reactions such as hives, difficulty in breathing, swelling (face, lips, tongue or throat), pale skin, necessitating drug discontinuation.
This inherent drawback of oral and transdermal rivastigmine tartrate administration warrants an alternative drug delivery system for rivastigmine tartrate. Hence in the present work an attempt is being made to provide an alternative colloidal drug delivery system for rivastigmine tartrate in the form of solid lipid nanoparticles which will have the following advantages
Sites specificity and controlled drug release.
Protection of drug against chemical degrada
High drug pay loa
Ease of manufacturing.
In the present work an attempt has been made to develop SLN of rivastigmine tartrate by micro-emulsification method and evaluate it for the following;
Preformulation studies on drug and polymer and to establish their compatibility in formulation using FT – IR.
To prepare solid lipid nanoparticles of rivastigmine tartrate.
Evaluation of the formulation for
Physical characterization of the solid lipid nanoparticles which includes
Particle size Analysis
Determination of Particle shape and Surface morphology
Percentage yield
Drug entrapment efficiency
In-vitro drug release study
Release kinetics
MATERIAL & METHODS
Rivastigmine tartrate was purchased from Swapnroop Drugs & Pharmaceuticals, Aurangabad, Maharashtra, India. Stearic acid was purchased from Loba Chemicals, Mumbai. Poloxamer 188 (BASF, Germany) Supplied by RFCL limited, Mumbai. All other chemicals and solvents used were of analytical grade.
Instrument Used
UV-Visible double beam spectrophotometer Shimadzu UV1800 with 1cm matched quartz cells. Electronic Balance. IR Spectrophotometer, Magnetic Stirrer, High speed propeller, Particle size analyser, Scanning Electron Microscope, Differential Scanning Colorimetry, Zeta potential
Preformulation Studies8
Preformulation testing is the first step in the rational development of dosage forms of the drug.
The goals of preformulation studies are
To establish its compatibility with different excipients.
To establishment the necessary physicochemical characteristic of a new drug
To determine its kinetic release rate profiles.
Hence, preformulation studies carried out with pure sample of drug include physical tests (description, melting point & solubility) and compatibility studies (drug with excipients).
Preparation of Calibration Curve
100 mg of rivastigmine tartrate was accurately weighed and dissolved in 100 ml water and methanol mixture (9:1) in volumetric flask, the resultant solution gives the concentration of 1mg/ml i.e.1000 µg/ml (stock solution-I). From this 10 ml solution was taken and then diluted up to 100 ml with the same solvent in a volumetric flask and then the concentration of this stock will be 100µg/ml (stock solution-II). From this stock solution-II10,20,30, 40, 50, 60, 70, 80, 90 and 100ml solutions were pippetted and volume was made to 100 ml using water to get concentrations of 10,20,30, 40, 50, 60, 70, 80, 90 and 100µg/ml respectively. The absorbance of these solutions was measured at 221 nm.
Compatibility Studies
A proper design and formulation of a dosage form requires considerations of the physical, chemical and biological characteristics of both drug and excipients used in fabrication of the product. Before producing the actual formulation, compatibility of rivastigmine tartrate with different polymers and other excipients were tested using the Infrared Spectroscopy (IR) technique and Differential Scanning Colorimetry (DSC).
FTIR Spectroscopy9
IR spectra of rivastigmine tartrate alone and along with excipients, this final complex were determined by Fourier Transform Infrared spectrophotometer using KBr dispersion method. The base line correction was done using dried potassium bromide. Then obtained mixtures were taken in a diffuse reflectance sampler and spectra were recorded by scanning in the wavelength region of 500 to 5000 cm−1 in a FTIR Spectrophotometer.
Differential Scanning Calorimetry (DSC) 10
DSC was performed in order to assess the thermo-tropic properties and thermal behaviour of the drug and the complex compacts prepared. A sample of 2-3mg was accurately weight was subjected to DSC run over the temperature range 40-350°C.
Preparation of Solid Lipid Nanoparticles13
The SLNs were prepared by Microemulsion based method. A 32 full factorial design was utilized in the present study for the development SLNs. Rivastigmine loaded SLNs were prepared from a warm o/w microemulsion containing Stearic acid as internal phase, poloxamer188 as surfactant and sodium taurocholate as co-surfactant. Microemulsion prepared by melting lipid (stearic acid) at 50ºC with measured quantity of drug, followed by sonication. To this poloxamer 188 was added and stirred for 2 min. Aqueous phase containing co-surfactant (sodium taurocholate) heated at 50ºC and added to melted lipid phase with mechanical stirring for 10 to 15 min, results in o/w microemulsion. This microemulsion was then added carefully dropwise into ice cold water present in a beaker with continuous stirring. Factors such as rate of addition, distance of needle from the surface of the beaker, rate of stirring were standardized to reduce particle size. In order to obtain optimum microemulsion, the needle was placed 4cm from the surface of the water and mixture stirred at 3000 rpm. The SLN dispersion was further stirred for 3hr after the complete addition of micro-emulsion. After completion of stirring, the SLN dispersion was subjected to ultra-sonication for a period of 10 min.
The nine batches (3x3) of SLN were prepared by varying the lipid concentration, surfactant concentration and co-surfactant concentration, using 32 factorial designs in three batches as shown in Table 1.
Table 1: Formulation Table of Rivastigmine Tartrate Loaded Solid Lipid Nanoparticles
Batch
Formn
Drug
(mg)
Stearic acid
(mg)
Poloxamer 188
(mg)
Sodium taurocholate
(mg)
Batch 1
F1
50
250
150
30
F2
50
500
150
45
F3
50
750
150
60
Batch 2
F4
50
250
225
45
F5
50
500
225
68
F6
50
750
225
90
Batch 3
F7
50
250
300
60
F8
50
500
300
90
F9
50
750
300
120
Evaluation and Characterization of the Prepared Solid Lipid Nanoparticles12-17
Percentage Yield

The practical percentage yield was calculated from the weight of solid lipid nanoparticles recovered from each batch in relation to the sum of the initial weight of starting materials. The percentage yield was calculated using the following formula.
Particle Size and Surface Morphology Analysis
Particle size analysis was done by using particle size analyser. Surface morphology was done by using Scanning Electron Microscopy (SEM).
Determination of Percentage Entrapment Efficiency
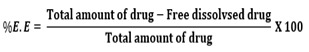
Entrapment efficiency of rivastigmine tartrate loaded solid lipid nanoparticles was estimated by centrifugation method. The prepared solid lipid nanoparticles were placed in centrifugation tube and centrifuged at 15000 rpm for 30 min. The supernatant (1ml) was withdrawn and diluted with water + methanol (9:1). The unentrapped rivastigmine tartrate was determined by UV spectrophotometer at 221 nm and calculated by following formula.
In vitro Drug Release Studies
Drug Release
In vitro dissolution studies were carried out in 900 ml of phosphate buffer 7.4 as a medium using USP apparatus type II (basket type). The rotation speed was 50 rpm and a temperature of 37±0.5˚C was maintained. The samples were analyzed by UV double beam spectrophotometer at λ 221 nm. Cumulative percentages of drug dissolved from solid lipid nanoparticles were calculated and graphs were plotted.
Release Kinetics
The data of in-vitro study was fitted in to three different kinetic models namely zero order kinetic model, first order kinetic model, Higuchi’s classical kinetic model. The mechanism of drug release is defined statistically in terms of co-relation co-efficient the highest values of co-relation co-efficient signify the particular release mechanism.
Zeta Potential
Zeta potential is an important and useful tool to indicate particle surface charge. Zeta potential was carried for all formulations of Rivastigmine SLNs.
RESULTS AND DISCUSSION
Preformulation Studies
The drug sample of rivastigmine tartrate was found to bewhite to off white powder having melting point 123 - 125°C and very soluble in water, soluble in ethanol and acetonitrile.
Compatibility Study
Physical Compatibility Study
Table 2: Result of drug excipients physical compatibility study after 15 days at 37ºC±2°C / 75%RH± 5 % RH
Sr. No.
Drug + Excipients
Initial Observation
After 15days at 37ºC±2°C / 75%RH ±5 %RH
1
Drug: Rivastigmine tartrate
White to off-white powder
Compatible
2
Stearic acid
A white to off white pellets
Compatible
3
Poloxamer 188
White to off white powder or solid prill
Compatible
4
Drug + Stearic acid
A white powder
Compatible
5
Drug +Stearic acid + poloxamer 188
A white to off white Creamy powder
Compatible
FTIR Compatibility Study
Figure 2: IR Spectra of Mixture of Rivastigmine Tartrate + Stearic Acid + Poloxamer 188
IR spectra of drug and polymer were obtained, which are depicted in Figure 2. All the characteristic peaks of rivastigmine tartrate were present in spectra at respective wavelengths (Table 3). Thus, indicating compatibility between drug and polymers. It shows that there was no significant change in the chemical integrity of the drug.
Table 3: Peaks (Cm-1) And Functional Groups Present – Rivastigmine Tartrate + Steric Acid + Poloxamer 188
Sr. No.
Peaks cm-1
Functional group
1
1597.06
C = C (Stre)
2
3172.90
C – H (Stre)
3
1849.73
C = O (Stre)
4
1294.24
C – O (Stre)
5
1544.98
Alkyl group
DSC Compatibility Study
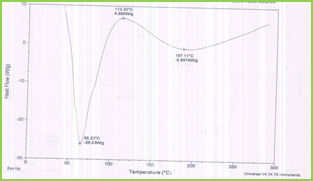
Figure 3: DSC Thermogram of Rivastigmine Tartrate + Stearic Acid + Poloxamer188
Figure 4: λmax of rivatigmine tartrate
The results of DSC analysis showed that the melting temperature for rivastigmine tartrate was found to be 113.30°C. The details of thermograms are shown in Figure 3. There was no significant changes observed.
Determination of λ max
The λ max of rivastigmine tartrate was determined in water and methanol mixture (9:1) which was scanned between 200-400nm in the UV spectrometer. It was found to be 221nm.
Standard Calibration Curve for Rivastigmine Tartrate
Calibration curve for rivastigmine tartrate was constructed using water + methanol (9:1) as solvent at 221nm.The concentration selected was 10 – 90 µg/ml (Table 4, Figure 5).
Table 4: Calibration data for rivastigmine tartrate
Concentration
(µg/ml)
Absorbance
(nm)
10
0.102
20
0.214
30
0.303
40
0.415
50
0.512
60
0.601
70
0.727
80
0.819
90
0.909
Figure 5: Standard Calibration Curve of Rivastigmine Tartrate
A straight line was obtained at R2=0.999. Equation of straight line was found to be y= 0.010x
Percentage Yield
The percentage yields of all nine formulations were calculated and were affected by concentration of polymer and the ratio of the mixture of polymers. The increase in polymer concentration leads to increase in percentage yield. The percentage yields of all formulations are shown in Table 5.
Table 5: Percentage Yield of Solid Lipid Nanoparticles of Rivastigmine Tartrate
Formulation code
Percentage yield (%)
F1
55.66
F2
66.36
F3
72.8
F4
54
F5
64.72
F6
70.37
F7
50.33
F8
60.36
F9
67.37
Particle Size Analysis
The mean particle size ranged from nm137 - 1300nm .The mean size was influenced by the concentration of lipid, surfactant and co-surfactant used in the formulations.
This may be due to the less availability of amphiphiles during emulsion formation and may be partly due to more partitioning of surfactant into oil phase as the concentrations of aqueous phase was increased. The particle size of SLNs decreases with increase in the concentration of poloxamer188. An increase in the concentration of sodium taurocholate leads to decrease the particle size of SLNs. Sodium taurocholate has the ability to decrease the size of the particles. Mean particle size of all formulations are given in the Table 6 and its graphical representation were shown in Figure 6 to Figure 14. The average mean particle size of all formulations were shown in Figure 15.
Table 6: Mean Particle Size and Polydispersity Index of Formulations
Formulation Code
Mean Particle Size (nm)
PDI
F1
1300
0.837
F2
194.7
0.980
F3
137.5
0.590
F4
531.0
0.938
F5
242.8
1.119
F6
212.2
1.279
F7
609.0
1.250
F8
191.0
0.890
F9
175.8
0.970
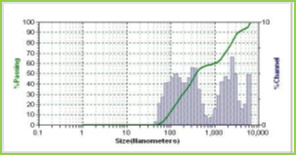
Figure 6
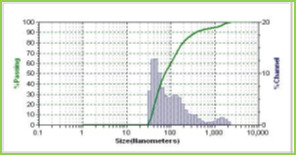
Figure 7

Figure 8
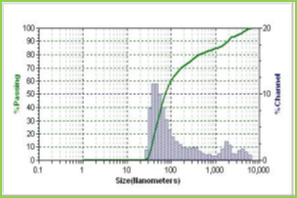
Figure 9

Figure 10

Figure 11

Figure 12
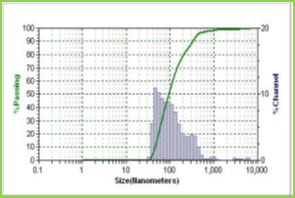
Figure 13
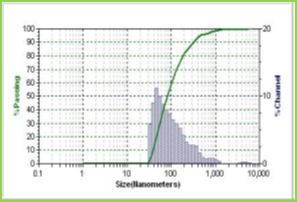
Figure 14
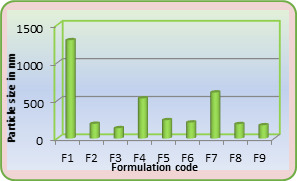
Figure 15: Average Particle Size

Figure 16: SEM Image of Rivastigmine Tartrate Loaded SLNs
Shape and surface Morphology
Solid lipid nanoparticles of rivastigmine tartrate were found to be spherical and irregular and their surface was smooth and devoid of cracks giving them good appearance. The SEM data obtained on the drug-loaded solid lipid nanoparticles of F9 shown in Figure 16.
Drug Entrapment Efficiency
The drug entrapment efficiency of a rivastigmine tartrate in sold lipid nanoparticles ranged from 93.26% to 99.80% (Table 7). It was observed that, when lipid concentration increased the entrapment efficiency was found to increase.
Table 7: Drug Entrapment Efficiency of Different SLN Formulations
Formulation code
Entrapment efficiency (%)
F1
93.26
F2
95.26
F3
97.82
F4
92.82
F5
96.04
F6
97.90
F7
93.35
F8
96.58
F9
99.80
Comparison of Formulations
Table 8 and Figure 17 shows comparison of % yield, % entrapment efficiency and particle size.
Table 8: Comparison of Percentage Yield, Drug Entrapment Efficiency, Particle Size of Solid Lipid Nanoparticles of Rivastigmine Tartrate
Formulation code
% Yield
% Drug entrapment efficiency
Particle size (nm)
F1
55.66
93.26
1300
F2
66.36
95.62
194.7
F3
72.87
97.82
137.5
F4
54
92.98
531.0
F5
64.72
96.04
242.8
F6
70.37
97.90
212.2
F7
50.33
93.35
609.0
F8
60.36
96.58
191.0
F9
67.37
99.80
175.8
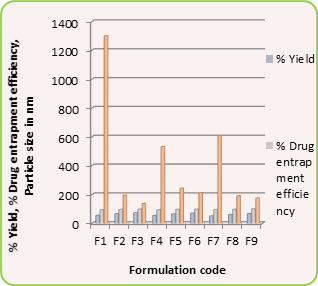
Figure 17: Comparison of % Yield, Particle Size and %Drug Entrapment Efficiency
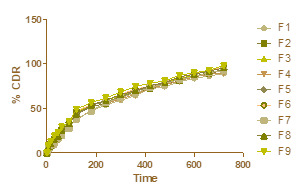
Figure 18: Zero order kinetic
In-vitro Drug Release
Release Kinetic Data for Solid Lipid Nanoparticle Formulations
The data obtained from in vitro drug release studies were fitted to zero-order, first-order and Higuchi’s equations and is represented in Figure 18, 19 and 20 respectively. After performing statistical analysis for release study data the coefficient of correlation was found to favour Higuchi’s classical diffusion model.

Figure 19: First order kinetic
Figure 20: Higuchi’s diffusion model
The values for regression coefficient shown in Table 9 for different kinetic models. From the results it is seen that the drug release mechanism from the formulation was found to follow Higuchi’s classical diffusion model. The rate of drug release is related to the rate of diffusion. The dissolution process is purely defined that the release rate is depends on the diffusion of drug from the lipid matrix, present in the developed formulation.
Zeta Potential
The zeta potential values obtained for the rivastigmine tartrate SLNs whichare given in Table 10 shows that the formulated rivastigmine tartrate SLNs are stable. F6 formulation was more stable than the other formulations.
Table 10: Zeta Potential of Rivastigmine Loaded Solid Lipid Nanoparticles
Formulation code
Zeta Potential(mV)
F1
-3.27
F2
-4.37
F3
-7.48
F4
-13.36
F5
-19.27
F6
-27.31
F7
-24.43
F8
-22.41
F9
-21.32
CONCLUSION
In the present work, solid lipid nanoparticles of rivastigmine tartrate were formulated to deliver rivastigmine in a controlled manner. A satisfactory attempt was made to develop solid lipid nanoparticles of rivastigmine tartrate and evaluated for in vitro characterization studies.
From the study following conclusions could be drawn.
Rivastigmine loaded SLNs were prepared successfully, and the process parameters were optimized using 32 factorial design.
The preformulation studies involving description, solubility, melting point of the drug were found to be comparable with the standard. Based on all the above preformulation studies, the drug rivastigmine tartrate was suitable for preparation of drug loaded solid lipid nanoparticles.
Drug-polymer compatibility studies by FT-IR and DSC gave confirmation about their purity and showed no interaction between the drug and selected polymers.
Practical and percentage yield increased as the concentration of lipid added increased.
Particle size studies revealed that mean size of the prepared SLNs was in the size range of 137nm -1300nm and particles were spherical & irregular in shape.
By varying the concentration of lipid, it was found that increase in lipid, surfactant (poloxamer188) and co-surfactant (sodium taurocholate) concentration in formulation leads to decrease in particle size, and increase in percentage entrapment efficiency and controlled release rate.
By performing in vitro drug release study it was observed that the drug release from the formulations increases as the particle size of the formulation decreases.
Rivastigmine tartrate release from all formulations followed Higuchi’s classical diffusion model kinetics.
Zeta Potential shown that the F1 formulation was more stable than others.
This outcome from release profiling strongly recommends that developed rivastigmine tartrate loaded solid lipid nanoparticles can be useful delivery carrier to deliver drug in controlled release manner.
REFERENCES
Jesus A, Ricardo I, and Joaquin R. Memory and neurogenesis in aging and Alzheimer’s disease. Aging and Disease. 2010; 1(1):30-36.
Russell S, Khan M. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Medical Hypotheses. 2004; 63:8–20. https://doi.org/10.1016/j.mehy.2003.12.045, PMid:15193340
Brasnjevic I, M. Harry, Steinbusch, Christoph S, Pilar M. Delivery of peptide and protein drugs over the blood–brain barrier. Progress in Neurobiology. 2009; 87:212–51. https://doi.org/10.1016/j.pneurobio.2008.12.002, PMid:19395337
http://www.tocris.com/dispprod.php?ItemId=330410#.U-m2ZvQW1SQ
http://www.mhra.gov.uk/home/groups/par/documents/websiteresources/con226931.pdf
http://dailymed.nlm.nih.gov/dailymed/lookup.cfm?setid=0e20124c-725a-0145-996c-2ff908703162
N. Basaran, Zelihagul D. Bioavailability file: Rivastigmine tartrate. Journal of Pharmaceutical Science. 2005; 30: 150-57.
Arthur I. Vogel. Elementary Practical Organic Chemistry. Part I: Small Scale Preparations. 2nd 76
Akhter S, Paul S, Ikramul H, Navid J, Syed S. and Reza S. Preparation, characterization and compatibility studies of naproxen loaded microspheres of cellulosic and polymethacrylic polymeric blend. Journal of Pharmaceutical Science. 2013; 12(1):11-21. https://doi.org/10.3329/dujps.v12i1.16295
Pintu K, Sahana B, and Soumen R. Enhancement of dissolution rate and stability study of ofloxacin solid dispersion. Der Pharmacia Sinica, 2011; 2(5):169-81.
Vijaya, D. Kalyana. Formulation and Evaluation of Solid Lipid Nanoparticles of Prednisolone. International Journal of Pharmaceutical Research. 2012; 4(1):73-76.
Gardouh A, Shadeed G, Ghonaim H. and Ghorab M. Preparation and characterization of glyceryl monostearate solidlipid nanoparticles by high shear homogenization. 1-26.
Akifuddin, Z. Abbas, Marihal S, A. Ranadev, I. Santosh and Dr. Kulkarni R. Preparation, characterization and in-vitro evaluation of microcapsules for controlled release of diltiazem hydrochloride by ionotropic gelation technique. Journal of Applied Pharmaceutical Science. 2013; 3(04):35-42.
Konwar R, Ahmad A. Nanoparticle: an overview of preparation, characterization and application. International Research Journal of Pharmacy. 2013; 4(4):47-57. https://doi.org/10.7897/2230-8407.04408
Silpa, R. Chakravarthi N, Yerram C, K. Hemant. Moxifloxacin loaded solid lipid nanoparticles (SLNs): Preparation and characterization. Asian Journal of Pharmacy and Research. 2012; 2(2) 105-12.
Shinde S. and Hosmani A. Preparation and evaluation lipid nanoparticles of fenofibrate obtained by spray drying technique. Pharmacophore. 2014; 5(1):85-93.
Khot V, Pillai M, Kininge P. Study of solid lipid nanoparticles as a carrier for bacoside. International Journal of Pharmacy and Biological Sciences.2013; 3(3): 414-26.
Read the full article
0 notes
Text
Samsung to showcase latest medical imaging & artificial intelligence for radiologists at RSNA 2018
Samsung to showcase latest medical imaging & artificial intelligence for radiologists at RSNA 2018
Samsung Electronics, a leader in medical imaging technology, is showcasing its latest ultrasound, digital radiography, computed tomography (CT), magnetic resonance imaging (MRI) concept and their software innovations at the Radiological Society of North America (RSNA) 2018 Annual Meeting at McCormick Place in Chicago.
"We are pleased that Samsung's AI technologies have been successfully applied to the existing diagnostic imaging devices and have been well received in the market," said Dongsoo Jun, president of health & medical equipment business at Samsung Electronics and CEO of Samsung Medison. "As a comprehensive diagnostic imaging solution provider, we will continue to strengthen our technologies through collaboration with hospitals and healthcare professionals. We aim to bring together radiologists and AI to fill in the gaps for improved healthcare management."
Samsung showcases several types of AI based diagnostic imaging software:
Ultrasound System -- S-Detect for Breast is AI based software which analyzes breast lesions using ultrasound images, and has been implemented into Samsung's ultrasound systems dedicated to Radiology. It assists in standardizing reports and classification of suspicious breast lesions by incorporating BIRADS ATLAS (Breast Imaging-Reporting and Data System, Atlas) According to a recently published study by radiology professor Tommaso Bartolotta from the University of Palermo in Italy, S-Detect for Breast has shown to assist various degrees of improvement in the overall diagnosis of breast lesions. When using S-Detect for Breast, the diagnostic accuracy for doctors with experience of four years or less improved from 0.83 to 0.87 (AUC, Area Under Curve). S-Detect for Breast could be a useful tool for supporting and guiding breast diagnosis among non-expert breast imaging physicians (Only shape and orientation descriptors are automatically classified in the United States).
Digital Radiography -- By using AI technology, the 'Bone Suppression' function, which reduces the bone signal from the chest x-ray image, clearly brings out the lung tissues obscured by the bones. 'SimGrid' has also been introduced as a solution to ease the workflow to replace grids, while providing excellent image quality with reduced scatter artifacts. According to a recently published study, 'SimGrid' provided image quality almost as good as the images taken with a physical grid. The Auto Lung Nodule Detection (ALND) is a CAD (Computer Aided Detection) solution based on AI technology used in the detection of lung nodules and is 510(k) pending. According to a recent study in press in Journal of Thoracic Imaging, ALND improved the detection sensitivity of lung cancer nodules 3 cm or smaller in chest radiographs by seven percentage points to 92 per cent, compared with the average of six chest radiologists.
Computed Tomography -- Samsung has introduced the intra-cranial hemorrhage package that combines a mobile stroke unit with a radiological computer aided triage and notification solution based on AI technology.
Magnetic Resonance Imaging -- Utilizing AI technology, Samsung is developing a technology to display information such as knee cartilage thickness as well as images of knee arthritis patients. The software is a prototype and not for sale in any region of the world.
At the Samsung booth, attendees will find a separate 'AI zone' that allows visitors to experience Samsung's AI technology more easily. Samsung will also host a symposium that will demonstrate how its image processing technology improves diagnostic accuracy and promote radiation safety for paediatric patients.
Read the full article
0 notes
Text
Do you know that "New study reveals probiotics do not help children with intestinal infections"
Do you know that "New study reveals probiotics do not help children with intestinal infections"
Probiotics are a multibillion-dollar industry with marketing claims of being an effective treatment for a multitude of ailments, including diarrhea. However, findings from a new study from the University of Calgary show the popular product has no effect on gastroenteritis, commonly yet erroneously called the stomach flu, in children.
"We studied the effects of giving probiotics to hundreds of children whose parents brought them into emergency departments across the country suffering from vomiting and diarrhea," says Dr. Stephen Freedman, MD, pediatric emergency medicine physician with Alberta Health Services, holder of the Alberta Children's Hospital Foundation Professorship in Child Health & Wellness and member of Cumming School of Medicine's Alberta Children's Hospital Research Institute. "We found no evidence that probiotics had any effect on reducing symptoms, or helping with recovery."
Freedman led a six-site Canadian study that included almost 900 children. He was also the co-principal investigator on a 10-site, concurrently conducted study in the United States, led by Dr. David Schnadower, MD that recruited almost 1,000 children. Findings from both studies will be published in the New England Journal of Medicine Thursday, November 22.
"There were smaller trials that had shown promising results. We wanted to replicate these findings on a large scale to see whether the age of the patient, the type of infection, and the use of antibiotics or length of time a child had the illness would affect the response to probiotics," says Schnadower, MD, who conducted the research as a professor of pediatrics at Washington University School of Medicine in St. Louis and is now a professor at University of Cincinnati College of Medicine. "The findings in both studies were consistently negative regardless of how the data were analyzed."
Researchers tested two commercially available brands of probiotics. Recruitment for the Canadian double-blind randomized study began in 2013. Children ranged in age from three to 48 months. Half of the children received probiotics while the other half received a placebo.
"These findings, taken together, are very powerful. The findings show that children treated with probiotics have the exact same outcomes across a large range of symptoms, as those given placebo -- the probiotics had no effect," says Freedman, who is also a member of the O'Brien Institute for Public Health at the Cumming School of Medicine. "The results deliver a clear message that we need to question the role and benefits of probiotics for other health applications using large, patient oriented, rigorous clinical trials."
Probiotics are generally considered safe to use. They are classified as food ingredients in Canada and can be sold as natural health products. As such, they do not require the same rigorous scientific testing that medications require, such as multiple clinical trials, in order to make beneficial claims. "Until now, most studies into the effects of probiotics have been small and industry funded," says Freedman. "In order to better serve families we need independent research to either prove or disprove the claims marketers are making on health care products."
Read the full article
0 notes
Text
Introducing New Youtube Channel - Subscribe Now
Read the full article
#fakeconferences#fakeconfernecesyoutubechannel#fakejournals#fakejournalsyoutubechannel#fakepublisher#fakepublishersyoutubechannel#JournalsClub#journalsclubYoutubeChannel#YoutubeChannel
0 notes
Text
Introducing New Youtube Channel - Subscribe Now
Read the full article
#fakeconferences#fakeconfernecesyoutubechannel#fakejournals#fakejournalsyoutubechannel#fakepublisher#fakepublishersyoutubechannel#JournalsClub#journalsclubYoutubeChannel#YoutubeChannel
0 notes