
The SBGrid Consortium is a Harvard Medical School-based software collaborative supporting the global structural biology community.
169 posts
Don't wanna be here? Send us removal request.
Last Seen Blogs

kapurb
kapurb

memyselfandlai
caroline

deepyogabeautyshoe
Untitled

normalessaints
NormaLessaints

dlhomiethigz
Untitled
Text
Using flies to study neurodegeneration
This publication highlight is part of the SBGrid/Meharry Medical College Communities Project, focused on science education and demonstrating how structural biology and preclinical science connect to medicine.
In the body, proteins carry out vital tasks to keep us alive. In order to do the wide variety of tasks needed to keep a living system going, proteins can form unique structures in order to carry out each unique function. These structures are encoded into the protein by its amino acid sequences, which are in turn determined by an mRNA sequence that is encoded by a DNA sequence. Information flows from DNA to RNA to protein structure in the cell and if any problems occur in this information pipeline, detrimental consequences follow. One example of a problem in the pipeline is a mutation in the DNA causing proteins to fold into the wrong shape and not be able to carry out their function. This is the case for Huntington’s Disease. In this neurodegenerative disease, the DNA that makes up the Huntington gene is mutated, almost resembling a skipping record, repeating the same nucleotide phrase, cytosine–adenine–guanine (CAG), over and over again. This repetition causes the resulting protein to have long regions unfolded or misfolded because of repeats of the glutamine amino acid (Gln, Q) and is referred to as a polyQ disease. Proteins with polyQ tracts are enriched in genes with neuronal function and typically aggregate together and cause the tragic symptoms that are hallmarks of the disease. In a recent publication in a IUCr Journal, SBGrid member Dr. Dierk Niessing, publishes a new protein structure that could help limit the aggregation of Huntington gene proteins.
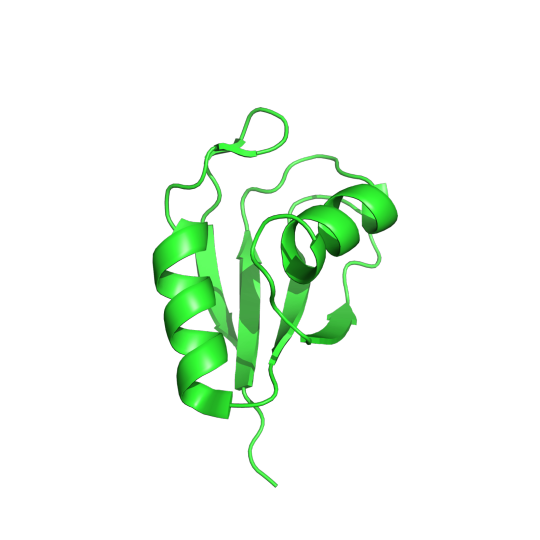
Pictured above is the determined structure of TRMT2A in Drosophila melanogaster. PDB:7PV5. CC BY SBGRID
Recent studies have shown that the inhibition of a protein called TRMT2A can lessen the aggregation of Huntington proteins, which means that TRMT2A could be a potential target for therapeutic molecules. If a drug can inhibit TRMT2A activity, it has the potential to help with Huntington’s disease. In order to determine how well this hypothesis works, we need a model organism that allows for ethical experimentation. Luckily, Dr. Dierk Niessing found a homolog of TRMT2A in the common fruit fly. He used protein crystallography and x-ray diffraction to observe the structure of this homologous protein. Upon examination, this fruit fly homolog had a different sequence than the human TRMT2A protein but a very similar structure. All the catalytic residues, the functionally relevant amino acids in the human protein, were also conserved in the fruit fly protein. This paper builds a case using structural biology that this fruit fly protein is a very similar homolog to the human TRMT2A protein. The implications of these findings mean that fruit flies could be an effective model to study how well drugs that target this protein are able to lessen Huntington symptoms. Read more about this fruit fly homolog in the Acta Crystallographica Section F.
-Vida Storm Robertson, Fisk University
Vida Storm Robertson is a Masters Student in Chemistry at Fisk University working in both solid-state and solution based structural determination techniques. He plans on starting a PhD program in biophysics in the fall of 2024.
0 notes
Text
Metals as nutrients for bacteria to thrive
This publication highlight is part of the SBGrid/Meharry Medical College Communities Project, focused on science education and demonstrating how structural biology and preclinical science connect to medicine.
Copper is a micronutrient that helps regulate many important functions needed for bacteria to survive. However, high amounts of copper can be toxic to bacteria so it’s important for bacteria to be able to maintain homeostasis. Copper’s main role is to act as a cofactor. A cofactor is a non-protein molecule that is crucial for a protein to carry out its function. Many proteins use copper as a molecular helper; because of this, copper is essential for all living things, yet, it is still unclear how bacteria get and maintain their copper levels. The work titled Stabilization of a Cu-binding site by a highly conserved tryptophan residue by SBGrid member Oriana Fisher looks into how the Gram-positive bacterium Bacillus subtilis regulates copper levels.
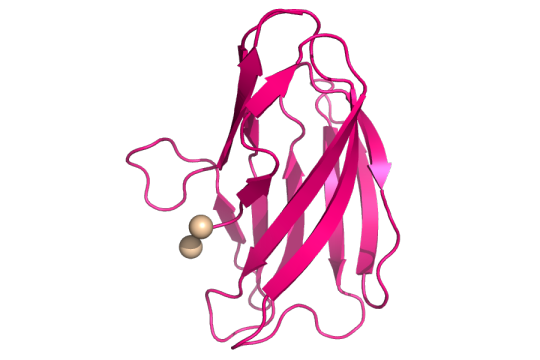
Structure of copper(II) (tan spheres) bound to the mutant YcnIW137F. CC BY SBGRID.
The authors looked at the ycnKJI operon that contains three copper-using proteins. An operon is a group of genes that are all controlled by the same promoter, or the place where genes are turned “on”. They found that a member of the ycnKJI operon, YcnI, a copper-binding protein whose function is still unknown, prefers to bind the oxidized version of copper. This oxidized version is known as Copper(II) oxide or Cu(II). By creating a mutated version of Ycnl where they swapped out a conserved tryptophan for phenylalanine, they found that even though tryptophan, an amino acid and binding site of Cu(II), is found in about 98% of this family of copper-binding proteins, it is not essential for Cu(II) binding. The conserved tryptophan, however, is necessary for maintaining the stability of Cu(II) binding to the protein.
Read more in the Journal of Inorganic Biochemistry.
- KeAndreya Morrison, Meharry Medical College

0 notes
Text
A small molecule that clogs up ribosome machinery
This publication highlight is part of the SBGrid/Meharry Medical College Communities Project, focused on science education and demonstrating how structural biology and preclinical science connect to medicine.
Ribosomes are essential molecular machines in the cell. They create proteins through the translation of RNA into an amino acid sequence. This process of translation is an important topic of study for molecular and structural biologists because of its vital role in maintaining life and the proteome. The production of the full proteome is essential for life and any variance or interference in the process could lead to drastic consequences. Because of this, the study of how small molecules and drugs interact and influence the ribosome is an ever-present question for structural biologists and medicinal chemists. In Nature Chemical Biology, SBGrid member Dr. Susan Shao from Harvard University, shows the unique effect that a small molecule, SRI-41315, has on the ribosome.
Ribosomes are made of many different protein and RNA units. The process of creating amino acid chains from an RNA transcript is highly dynamic and requires the timely introduction and removal of a library of participating molecules in the translation symphony. One of these molecular players in translation is eukaryotic translation termination factor 1, eRF1. This protein binds to the ribosome subunits and aids in the termination or conclusion of the translation process. eRF1 helps the ribosome read the translation instruction, the mRNA, and helps the ribosome know when to stop and remove the finished amino acid chain. The small molecule SRI-41315 has been known to degrade eRF1, but the mechanism for this degradation was unknown until Dr. Susan Shao and colleagues proposed the following using cryo-EM and molecular assays:
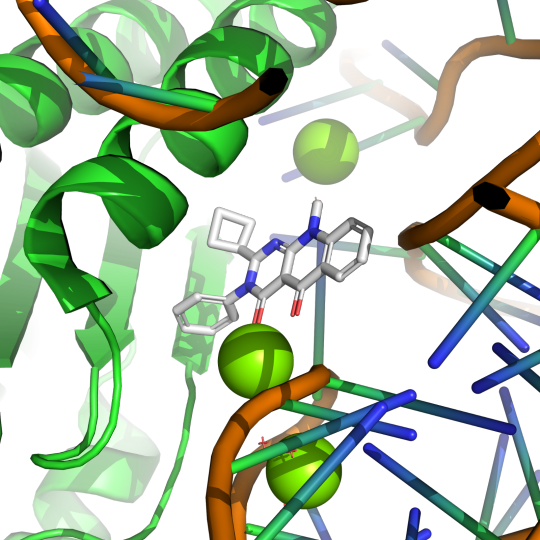
Pictured above is small molecule SRI-41315 (gray) interaction with eRF1 (green) and RNA (orange) in the ribosomal complex. PDB:8SCB. CC BY SBGRID
Using electron microscopy at cryogenic temperatures, a technique known as cryo-EM, Dr. Shao was able to create a model of eRF1 in complex with a ribosome with SRI-41315 bound. In this model, SRI-41315 was shown to act as a glue in the machine. This small molecule locked eRF1 into a specific configuration, limiting the dynamic process of translation. By gumming up the ribosomal machine, SRI-41315 increased eRF1 degradation and affected the machine's ability to accurately produce functional proteins. The ability of this small molecule to indirectly slow down and effect the efficiency of translation is a very compelling observation and provides evidence for the wide array of functions small molecules can have in living systems.
To learn more about the clog in the ribosomal machine, continue reading at Nature Chemical Biology.
Vida Storm Robertson, Fisk University
Vida Storm Robertson is a Masters Student in Chemistry at Fisk University working in both solid-state and solution based structural determination techniques. He plans on starting a PhD program in biophysics in the fall of 2024.
1 note
·
View note
Text
An inside look into the Powerhouse of the Cell
This publication highlight is part of the SBGrid/Meharry Medical College Communities Project, focused on science education and demonstrating how structural biology and preclinical science connect to medicine.
Mitochondria are the powerhouses of the cell. We’ve all heard this phrase at some point in our lives, but what does it mean on a molecular level? A mitochondrion is an organelle in cells like animals, plants and humans that functions to provide the cell with energy in the form of a molecule called adenosine triphosphate, or ATP. A sufficient stock of ATP is produced in mitochondria when cells in our body convert food into energy, or what is known as cellular respiration. More specifically, cellular respiration is when our cells take in glucose and oxygen through many steps and then release carbon dioxide, water, and energy. An important step is to store this energy into a form that can be used in different parts of the cell when needed. This is done by moving electrons (really small particles that are a part of atoms) by a group of biomolecules called proteins. These proteins come together to form an electron transport chain. As the name suggests, the electron transport chain is a series of processes where electrons are transported from one molecule (donor) to another molecule (acceptor) resulting in the formation of ATP. For the electron transport chain to work, it relies on electron donor molecules produced from the Krebs cycle, also known as the citric acid cycle.
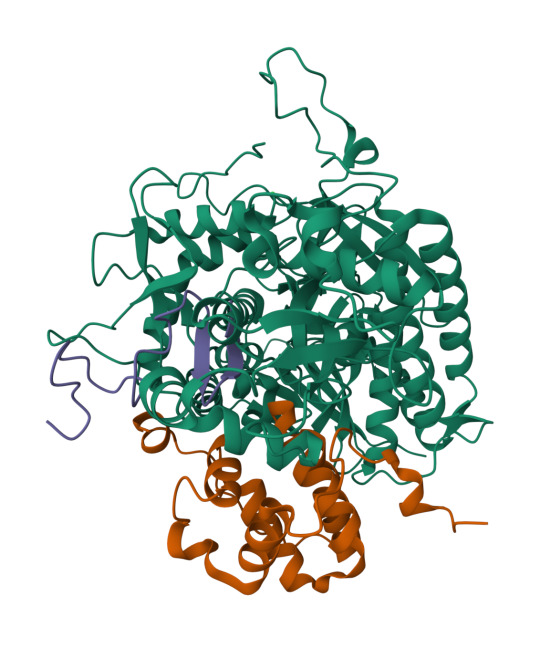
The Krebs cycle is a metabolic pathway in the mitochondria that allows our bodies to break down carbohydrates to be used for energy. This process starts with pyruvate entering the system and, after several steps, ends with the production of several molecules such as, ATP, NADH, and FADH2. These important molecules enter the electron transport chain acting as electron donors to make more ATP. CO2 is also produced from the Krebs cycle and enters other metabolic pathways. All of the steps in the Krebs cycle are carried out by different enzymes. One of the enzymes is succinate dehydrogenase (SDH). As a member of the electron transport chain in mitochondria, SDH or complex II, catalyzes the formation of fumarate from succinate- releasing FADH2 as a byproduct. SDH produces a fraction of the total ATP, but is important for a wide range of cellular activities, such as suppressing tumor growth and neurological development. Although the importance of SDH has been widely established, what is unknown are the details of how the regulation of SDH assembly factors (SDHAF) and SDH assemble into a functional unit.
Left: structure of SDHA-SDHAF2-SDHAF4 assembly intermediate (PDB: 8DYD). Green: SDH, purple: SDHAF4, orange: SDHAF2.
Right: structure of SDHA-SDHAF4 assembly intermediate (PDB: 8DYE). Green: SDH, orange: SDHAF4. CC BY SBGRID.
To address this question, SBGrid member Tina Iverson investigated the intermediate complexes formed by the SDHAF subunits and cofactors. Through the use of cellular studies and biochemical experiments, along with X-ray crystallography and NMR for structural studies, the authors were able to map out the different intermediate complexes that lead to the formation of the fully matured SDH. Interestingly, the authors found that regions of the assembly proteins can transition from ordered (a protein with a secondary structure) to disordered (a protein with no secondary structure) and that this change is important for the transfer between intermediate complexes. These results shed light on the previously unknown steps of the formation of a mature and functional complex II of the electron transport chain.
Read more in Nature Communications.
KeAndreya Morrison, Meharry Medical College

0 notes
Text
Do you know what can cause acne?
This publication highlight is part of the SBGrid/Meharry Medical College Communities Project, focused on science education and demonstrating how structural biology and preclinical science connect to medicine.
Everyone will have some experience with acne in their lifetime. Generally, most people struggle with acne during their teenage and young adult years, but acne can persist in some people during their adult years. Acne is a condition where pores and hair follicles in the skin get clogged. Pores can be clogged with dead skin cells, sebum (an oily substance that provides a barrier for the skin), and bacteria. Clogged pores create pimples that can swell and become painful. Cutibacterium acnes (C. acnes) is a common bacterium that plays a role in the health of our skin . It is the bacteria most commonly associated with acne even though it is found on healthy and acne-prone skin similarly. Like many other bacteria, C. acnes has multiple strains. Some strains are associated with acne-prone skin while others are associated with healthy skin. However, not much is known about the genetic factors that make a strain of C. acnes acne-causing. One key difference between C. acnes strains that promote healthy is the variation in the enzyme hyaluronidase. There are several television commercials that mention acne treatments that contain hyaluronidase. Hyaluronidase is an enzyme that breaks down hyaluronic acid. C. acnes can express two variants of hyaluronidases, HylA and HylB. HylA and HylB are about 74% similar. HylA is associated with acne-prone skin while Hyl B is associated with healthy skin.
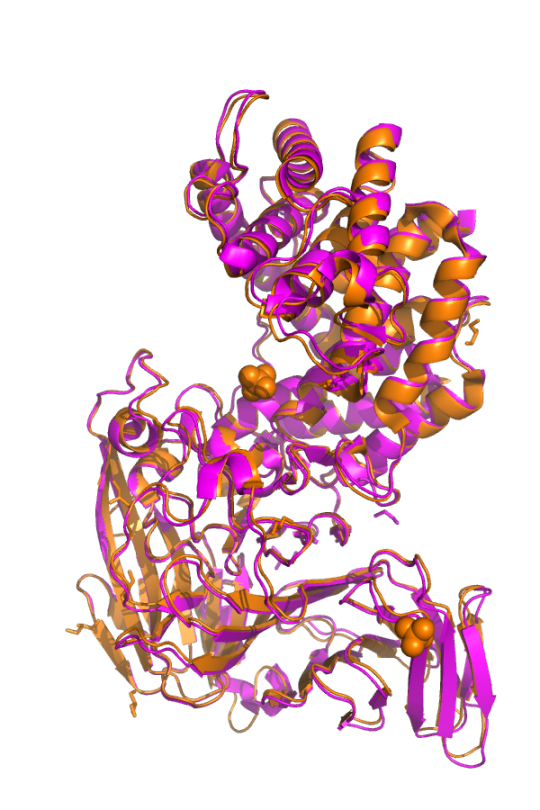
Pictured above is HylA (pink; PDB: 8FYG) overlayed with HylB (orange; PDB: 8FNX). CC BY SBGRID.
In this work, titled Functional divergence of a bacterial enzyme promotes healthy or acneic skin, SBGrid member Ramachandran Murali and colleagues uncover the different mechanisms of action between HylA and HylB that contribute to acne or healthy skin, respectively. HylA breaks down hyaluronic acid into large fragments. These large fragments help promote an inflammatory pathology that leads to acne. Contrastingly, HylB degrades hyaluronic acid solely into disaccharides (two sugar molecules) that lead to reduced acne. Along with solving x-ray crystal structures of different mutants of HylA and the wildtype of HylB, the authors also show the potential for targeting HylA using inhibitors and peptide vaccines. Their findings suggest that inhibiting HylA could be a potential target for future acne treatments.
Read more in Nature Communications.
- KeAndreya Morrison, Meharry Medical College

0 notes
Text
New SARS-CoV-2 antibody generated to bind to multiple conformational states of spike proteins.
This publication highlight is part of the SBGrid/Meharry Medical College Communities Project, focused on science education and demonstrating how structural biology and preclinical science connect to medicine.
The emergence of SARS-CoV-2 in 2019 created a worldwide pandemic that is still present in the population today. Scientists like Dr. Jason McLellan and Dr. Kizzmekia Corbett have published methods for isolating stable versions of SARS-CoV-2 spike proteins that can be studied in a research lab. Structural biologists and pharmacologists have created many effective treatments and vaccines for this respiratory virus, but the efficiency of these therapies changes as the virus mutates. The high viral load of SARS-CoV-2 in the population allows for constant evolution of the virus, specifically selecting for mutates able to avoid prevalent treatments. Thus, it is no surprise that the latest variant, Omicron, showed a unique ability to avoid immune detection and recognition from formerly designed antibodies. Drs. Jason McLellan and Andrew Ellington, in their latest paper in Communication Biology, show a unique method of developing new antibodies and describe how this newly developed antibody performs against the most prevalent variants of SARS-CoV-2.

Pictured above is the receptor binding domain of COVID-19 (green) binding to antibody N3-1 (blue). PDB:6M0J. CC BY SBGRID
New variants of SARS-CoV-2 are able to avoid the immune system's detection and current antibody binding because of mutation and variations in dynamics. To maintain the effectiveness of current COVID-19 treatments new antibodies must be designed to bind to these emerging mutates. In this paper, new antibodies that preferentially bind to the spike protein of new SARS-CoV-2 mutates are designed using an integrative cellular and structural approach. Antibodies are made up of two interchangeable parts, a heavy chain and a light chain. The heavy chains of antibodies created in this paper were taken from serum donated by patients who had previously contracted COVID-19. These patient-derived heavy chains were combined with light chains that were previously published in public data banks. Combinations of heavy and light chains were tested for efficiency of binding to SARS-CoV-2 spike protein. A novel antibody created using the methodology above showed high efficiency. This unique antibody's high binding was due to its ability to bind to many different conformations of the spike protein. This ability to bind to the SARS-CoV-2 spike protein, in spite of its protein dynamics, allows this new antibody to be much more effective at targeting more evasive strands of the virus.
To read more about SARS-CoV-2 and this new method of antibody development, check out the paper in Communications Biology.
- Vida Storm Robertson, Fisk University
Vida Storm Robertson is a Masters Student in Chemistry at Fisk University working in both solid-state and solution based structural determination techniques. He plans on starting a PhD program in biophysics in the fall of 2024.
0 notes
Text
Designing Amphotericin B derivatives to be less renal toxic
This publication highlight is part of the SBGrid/Meharry Medical College Communities Project, focused on science education and demonstrating how structural biology and preclinical science connect to medicine.
Amphotericin B is a highly effective, highly toxic antifungal medication that is regularly used to treat serious infections in clinical settings. Amphotericin B removes key lipids from both human and pathogen membranes, causing the cell death of infecting organisms but with very concerning consequences to patients. For this reason, amphotericin B has been used only in severe circumstances to spare patients from its cytotoxicity. SBGrid member Dr. Chad Rienstra at the University of Wisconsin-Madison and colleagues used amphotericin B as the backbone to rationally design an antifungal able to selectively interact with fungus membranes and limit interactions with patient tissues.
In a new Nature publication, Dr. Chad Rienstra used solid-state structures using nuclear-magnetic resonance of amphotericin B bound to lipids in cellular membranes. Amphotericin B had a preference to bind strongly to critical membrane lipids called sterols. Dr. Rienstra showed NMR structures for amphotericin B bound to two key types of sterols, cholesterol and ergosterol. Cholesterol is very common in human membranes and ergosterol is found exclusively in fungi membranes. The NMR structures showed different mechanisms of interaction between amphotericin B and its two sterol ligands. Using these structural differences, modifications were introduced to lower sterol interaction making the drug less renal-toxic. This difference in reactants allowed the design of amphotericin B based molecules that selectively interact with ergosterol and do not interact with cholesterol. This small-molecule tuning based on natural products allowed researchers to develop a promising new antimicrobial agent that targets key chemical differences between host and pathogen membranes. These derivatives are currently being studied in clinical trials. Dr. Marty Burke, a key collaborator and fellow corresponding author at the University of Illinois Urbana-Champaign, talks about this paper on the Nature Podcast here.
Read more in Nature.
-Vida Storm Robertson, Fisk University
Vida Storm Robertson is a Masters Student in Chemistry at Fisk University working in both solid-state and solution based structural determination techniques. He plans on starting a PhD program in biophysics in the fall of 2024.
0 notes
Text
New roles for the histone methyltransferase MLL1
This publication highlight is part of the SBGrid/Meharry Medical College Communities Project, focused on science education and demonstrating how structural biology and preclinical science connect to medicine.
Epigenetics is the study of all the things that influence how our genes work that do not involve changes in our DNA sequence. Epigenetics can include behavioral factors, such as eating and drinking habits, and environmental factors, such as living in a region with warmer versus colder temperatures. These factors impact how our genes are expressed by influencing biological processes like DNA methylation and histone modification. DNA methylation occurs when a chemical known as a methyl group is added to specific places on DNA strands to stop a gene from being transcribed or “read”; this process is usually referred to as turning a gene “off.” Histone modification refers to how proteins called histones are altered. DNA wraps around histones. When they are tightly wrapped, DNA expression is turned “off” and when they are wrapped loosely or not at all, DNA expression is turned “on.” Chemical groups, such as methyl groups, can be attached to histones to help regulate how tightly the DNA is wrapped around it. These two processes, DNA methylation, and histone modification, can be altered by many different environmental factors that affect our health. When epigenetic regulators are negatively altered it is known as epigenetic dysregulation. Epigenetic dysregulations can be inherited, but it is often reversible through treatment. Epigenetic dysregulation is also a hallmark of cancer since altered gene expression usually favors the growth and progression of tumor cells.

Pictured above is the Co-crystal structure of MLL1 SET domain (green) with Borealin (blue) (PBD: 7U5V). CC BY SBGID.
A study published by Yali Dou along with SBGrid member Uhn-Soo Cho, titled Non-canonical MLL1 activity regulates centromeric phase separation and genome stability, looks at MLL/KMT2, a family of histone methyltransferases. Histone methyltransferases are enzymes that add methyl groups to histones. The normal functions of MLL/KMT2 have been well documented in the past, especially their role in cancers. However, their abnormal, or non-canonical, functions that affect genetic stability are not as well explored. The authors discovered that MLL1 methylates Borealin K143, a chromosomal passenger protein that helps regulate cell division. They went on to further show that methylation of Borealin K143 by MLL1 regulates liquid-liquid phase separation. Liquid-liquid phase separation is important for chromosomal passenger protein localization at centromeres, a region of chromosomes that is important for helping a cell divide its DNA correctly during cell division. They also solved a crystal structure of MLL1 in complex with Borealin K143. These findings suggest that targeting MLL1 in hepatocellular carcinoma patients who have chromosomal instability could be an effective option for treatment.
Read more in Nature.
- KeAndreya Morrison, Meharry Medical College

0 notes
Text
Structure of Equity - Jamaine Davis - Meharry Medical College
Sharing some of our #SBGrid member tales from the last year. This one from September 2022.
Numbers speak clearly to Jamaine Davis. As a boy growing up on Long Island, math came so easy to him that one of his family nicknames was "the professor."

Other numbers have shaped his ambitions at Meharry Medical College in Nashville, Tennessee, where Davis runs one of the few labs in the world that uses structural biology to help explain biological health disparities.
For example, U.S. Black adults are twice as likely to have Alzheimer's disease compared to non-Hispanic Whites. And despite a somewhat lower overall lifetime risk of breast cancer, Black women experience a 40% higher death rate from breast cancer than White women at every age and are more likely to be diagnosed with fast growing and late-stage breast cancer.
"My research program is basically at the intersection of structural biology, genetics and disease, and health disparities," says Davis of the big-picture questions that guide his lab's work. "What are the molecular mechanisms that dictate who develops diseases like cancer or Alzheimer's? And then how do we design effective therapies? How do we target the right pathways for the right treatment for that patient?"
One project in the early stages focuses on a gene (ABACA7) that has a stronger effect on risk of Alzheimer's disease in Blacks than the better known ApoE4 gene risk variant. "It's actually the strongest risk factor for developing Alzheimer's in African Americans known so far," Davis says.
As he explains it, ABACA7 transports lipids out of cells, handing off the lipids directly to ApoE, and also interacts with Tau, another protein that goes awry in Alzheimer's. Two missense variants in ABACA7 confer the risk.
"So we've been studying these mutations to see what impact they have on lipid transport," Davis says. "Once we're done, we can look at the people who particularly carry this mutation or variant, see what downstream processes are altered, and design therapies to rescue that. And these variants so far have only been identified in African Americans."
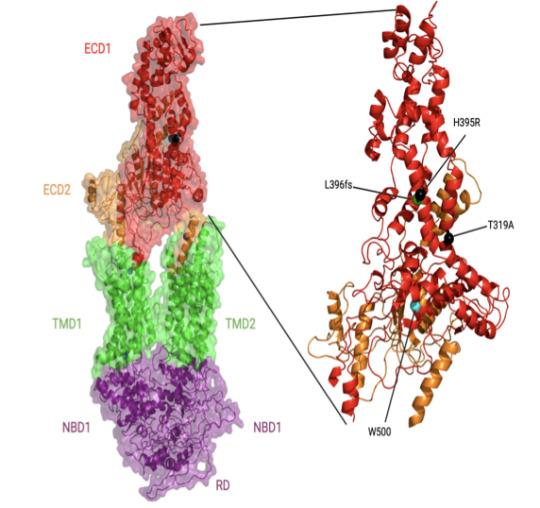
In individuals with African ancestry, the phospholipid-transporting ATPase ABCA7 (ABCA7) gene has stronger associations with Alzheimer’s disease risk than in individuals with European ancestry and than the apolipoprotein E (APOE) ɛ4 allele. The Davis lab is exploring the structure and function of key ABACA7 mutations and how they contribute to alterations in transporting lipids, which may influence Alzheimer’s in African Americans. Credit: Courtesy of J.Davis.
Davis began his academic training on a different career path. With his early affinity for math and science, he reasoned that chemical engineering made sense as a college major. But near graduation at Drexel University, he realized that the typical next step for someone with a chemical engineering degree was a job at an oil company. He hadn't taken one biology course in college, but he found himself drawn to biomedical research instead.
He seized an opportunity to work in a biophysics lab at a neighboring school, University of Pennsylvania, where his mentor Jacqueline Tanaka gave him a peek at the scientific career he could have in biophysics and opened his eyes to the kind of academic role model he could be. Her excitement for X-ray crystallography and for increasing the proportion of women and minorities in science inspired him to go to graduate school.
"She built her career in structural biology and mentoring, hand in hand," Davis says. "She saw some potential in me, and I was at a crossroads." Davis had also been unaware of the extent of health inequities across the country and of the low representation of minorities in academia.
For his thesis, Davis chose the lab of Harvey Rubin, a dynamic speaker who fostered an immediate interest in infectious disease. In Rubin's lab, Davis characterized an enzyme that enables Mycobacterium tuberculosis to enter (and possibly exit) the dormancy stage in the lungs of people.
When Davis finished his PhD in 2007, he was the first Black to earn a doctorate in biochemistry and molecular biophysics at UPenn. "I had a great time," he says. "They were very supportive. But it is pretty shocking. If you look at Twitter, there are other people posting the same kind of statistic. They're the first Black to graduate from a certain program at a certain institution. It does show there is still some under-representation across different departments."
He followed up with two postdoctoral fellowships at the National Cancer Institute. He first showed that a novel protein in Shigella (bacteria that cause food poisoning) was not a protease, as some suspected. A second project elucidated the binding modes of a protein with multiple domain repeats implicated in the development of cancer.
Then he thought about how best to combine his interests in a distinctive research program. He chose Meharry, one of the oldest and largest historically black U.S. academic health centers. (Davis is also a member of the Vanderbilt University Center for Structural Biology.)
Historically black colleges and universities are powerhouses in educating African Americans who go on to earn doctoral degrees in science, technology, engineering, math, and medicine, as Davis and his co-authors reviewed in a commentary (Cell, 2022). Blacks make up 12% of the U.S. workforce, but only 5% of working physicians and 3.6% of full-time faculty conducting research at medical schools.
When the COVID-19 pandemic hit, Davis found new opportunities for mentorship and community outreach. Soon after the pandemic took hold, a student-driven community formed on Twitter with the handle @BlackInBiophys and a logo designed by Taneisha Gillyard, a former postdoc in the Davis lab. Davis spoke at a virtual meeting held by the group.

A former postdoc in the Davis lab, Taneisha Gillyard, designed the logo for @BlackInBiophys, a student-driven community that formed on Twitter during the pandemic. Credit: Taneisha Gillyard.
In a short time, a strong sense of community developed among people who may have never met in person, but know a lot more about each other through social media, Davis says. People share grant writing tips, training and job opportunities, and generally celebrate the scientists, their contributions, and career options for the next generation.
The visibility may help change other statistics about Black researchers receiving less NIH funding and being cited less often than their white colleagues, Davis says.
Davis also teamed up with Meharry colleague Jennifer Cunningham-Erves to develop a funded community outreach project to address community concerns about vaccines. He has spoken about the basic science of mRNA at townhall-style community meetings, in person and virtual. The online recordings have reached people from Chicago to New York to Haiti.
The project collaborates with a consortium of more than 90 churches in middle Tennessee and Better Options TN, a community nonprofit organization. To understand concerns, the Meharry team interviewed people in the Southern United States. They developed and organized content on a frequently updated web site, https://yourcovidvaxfacts.com/en.
"We asked about their thoughts about the vaccine and the virus," Davis says. "The biggest one, particularly for Black Americans, was the distrust with government and healthcare. But I was very impressed with some of the questions that the public had. They weren't getting answers, and they wanted answers. If you remember, one of the major issues with people not getting a vaccine was that they thought it would affect their DNA. They just weren't familiar with mRNA."
Davis felt their concerns and trust issues as well. He initially was cautious about being vaccinated himself, waiting to see more data about its safety in people. "Even being a scientist, I was hesitant," he says. "I didn't want to be one of the first," he says. But as he explained the science and helped alleviate concerns of others, he also convinced himself to get the vaccine too.
Meanwhile, back in the lab after the pandemic disruptions, Davis and his team are working to improve health outcomes for populations most at risk, one variant protein and pathway at a time.
- Carol Cruzon Morton
0 notes
Text
Novel high throughput assay development for cancer inhibitors
This publication highlight is part of the SBGrid/Meharry Medical College Communities Project, focused on science education and demonstrating how structural biology and preclinical science connect to medicine.
High through-put screens (HTS) are experimental investigation tools that use a variety of automated equipment to run experiments (sometimes called assays). HTS are especially important in the field of drug discovery where it is a key process in identifying “hits.” Hits are the compounds/drugs that show promising results in the early phases of drug development that are then chosen for more specialized testing. HTS allow testing of thousands, sometimes even millions, of these compounds in a short time frame. Screening all of those possible compounds individually could take a person months or maybe even years. HTS not only significantly decrease the time it takes to run assays, but they also reduce the risk of human errors.
While many HTS are available, none aim at finding an inhibitor compound for the PARP1-HPF1 complex. The PARP1-HPF1 complex stands for Poly (ADP-ribose) polymerase 1, an important enzyme in the DNA damage response cascade, and Histone PARylation Factor 1, a regulator of PARP1. This complex is integral for repairing DNA damage in cells and inhibition of the complex ultimately leads to cell death. The PARP1-HPF1 complex exhibits the same DNA damage repairing mechanisms as well as inhibition of cell death in tumor cells. Several PARP inhibitors have been developed to treat ovarian cancer, however, about 40% of patients with ovarian cancer fail to respond to PARP inhibitors and many patients develop a resistance to them after repeated doses. None of the current inhibitors target the PARP1-HPF1 complex, which researchers think may be more advantageous for cancer drugs since targeting this complex will allow for more specificity and inhibit functions of PARP1 that are only observed with HPF1 and not PARP1 by itself. Currently there are no HTS to identify hits for inhibitors of the PARP1-HPF1 complex.

SBGrid member Karolin Luger and colleagues have developed a HTS that will identify hit compounds for inhibiting the PARP1-HPF1 complex. Their HTS is based on a fluorescence polarization assay that measures the activity of PARP1. In short, the complex is incubated with potential inhibitors and the PARylation reaction is initiated by the addition of NAD+. The fluorescence signal is then measured. A low fluorescence signal indicates that there is a disruption of the complex and a high signal indicates that the complex is maintained and also inhibited. This assay was thoroughly tested and scaled to analyze 10,000 compounds at a time. Hopefully, the use of this novel HTS will help to identify key compounds that will help us further study PARP1/HPF1 interactions in vitro and in cellular models of cancer along with finding new inhibitors to develop more effective cancer drugs.
Read more in SLAS Discovery.
KeAndreya Morrison, Meharry Medical College

0 notes
Text
Using MicroED for molecule conformations
This publication highlight is part of the SBGrid/Meharry Medical College Communities Project, focused on science education and demonstrating how structural biology and preclinical science connect to medicine.
Microcrystal electron diffraction (MicroED) is an emerging technique that has been shown to determine the solid-state structure of small molecules and small proteins in microcrystals. The ability to gain structural insight from microcrystals allows for a lower burden on crystallographers and opens a new range of target molecules that were previously unavailable due to their inability to form high-quality crystals. One such molecule is the commonly prescribed drug Mirabegron. Mirabegron is used to treat overactive bladder symptoms, yet a high quality structure of the molecule has eluded scientists due to its powdery crystalline state. SBGrid member Dr. Tamir Gonen, from UCLA, was able to determine a structure of Mirabegron using MicroED and sample a stable conformer state of this important drug.

Pictured above is Dog beta3 adrenergic receptor (grey) and Mirabegron (cyan). Mirabegron is shown deeply embedded in the binding site of this adrenergic receptor. PDB:7DH5. CC BY SBGRID
In a study published last month in Advanced Science, Prof. Gonen was able to show that Mirabegron has two distinct conformer states, a cis and trans, in a low energy crystal. This lattice unit is held together using a myriad of interactions between Mirabegron molecules, but a majority of the attraction is due to the demand to bury hydrophobic regions. While this arrangement is low energy and highly stable, it does not appear to be the conformer state found upon binding to target sites in proteins. The image above shows that Mirabegron buries deep into binding sites on proteins, and to do this it would need to go through a large conformational change from the states sampled in MicroED. These insights help to push forward our knowledge of the Mirabegron mechanism of action and could help describe the off-target effects of this drug.
- Vida Storm Robertson, Fisk University
Vida Storm Robertson is a Masters Student in Chemistry at Fisk University working in both solid-state and solution based structural determination techniques. He plans on starting a PhD program in biophysics next fall.
0 notes
Text
A look into specific interactions between Bruton’s Tyrosine Kinase (BTK) and its inhibitors
Note: This publication highlight is part of the SBGrid/Meharry Medical College Communities Project, focused on science education and demonstrating how structural biology and preclinical science connect to medicine.
Non-receptor tyrosine kinases (nRTK) are a subgroup of tyrosine kinases that are responsible for the phosphorylation of proteins. As the name suggests, tyrosine kinases work by transferring a phosphate group from ATP to the tyrosine residue of a protein. Non-receptor refers to the group of tyrosine kinases found within the cytosol of the cell, unlike receptor kinases, which are embedded into the cellular membrane. nRTKs are involved in many cell functions such as regulating cell growth and proliferation. They also play critical roles in immune system regulation. One specific nRTK that is involved in propagating signals from B cell receptors is known as Bruton’s tyrosine kinase (BTK). BTK’s importance was discovered when it was revealed that mutations in the Btk gene, the gene that encodes BTK, leads to the development of X-linked agammaglobulinemia (XLA), an immunodeficiency disease. BTK also plays essential roles in many diseases such as mantle cell lymphoma, chronic lymphocytic leukemia, Waldenstrom macroglobulinemia, small lymphocytic lymphoma, marginal zone lymphoma, and chronic graft-versus-host disease, just to name a few. These factors combined have made BTK inhibition a target of several drug therapies aimed at treating B cell malignancies. These therapies include the first in class BTK inhibitor, Ibrutinib, and the second-generation inhibitors Acalabrutinib, Zanubrutinib and Orelabrutinib. Although all of these therapeutics have seen success in clinical applications, specific interactions between the drugs and BTK are not well understood.

Above: Front and back views of BTK/Acalabrutinib complex (PDB: 8FD9) CC BY SBGRID.
In this work, SBGrid member Amy Andreotti and colleague David Lin reported the first structure of BTK in complex with Acalabrutinib. They also report a structure of BTK with Tirabrutinib, another second-gen BTK inhibitor that, at the time of this publication, is in clinical use in Japan and Taiwan but not yet FDA approved. When comparing their BTK/Acalabrutinib complex structure with a previously reported structure of BTK/Ibrutinib complex, the authors noted several regions where structural differences occur based on evaluation of RMSD values. Comparisons between BTK/Acalabrutinib and BTK/Tirabrutinib reveal large conformational differences in the activation loop that the authors attribute to the kinase undergoing dynamic fluctuations when bound to the drug, after further investigation of the previously reported structure of BTK/Tirabrutinib complex. Along with broad structure characterization, the authors also identified a few key residues that interact with the inhibitors. Combined, their work shows the need for further probing into how these drugs interact with BTK in order to fully examine how these inhibitors bind.
Read more in PLOS One
- KeAndreya Morrison, Meharry Medical College

1 note
·
View note
Text
A personal history of the great Crystallography pioneer Dr. Wayne Hendrickson
Note: This publication highlight is part of the SBGrid/Meharry Medical College Communities Project, focused on science education and demonstrating how structural biology and preclinical science connect to medicine.
SBGrid member Dr. Janet L. Smith writes a beautiful history of crystallography legend and pioneering physicist Wayne Hendrickson in the International Union of Crystallography Journal. In this commentary, Dr. Smith gives a wonderful history of Dr. Hendrickson’s academic work as he developed into the world class researcher we all know today. Hendricks recently won the Ewald Prize from the International Union of Crystallography and published a magnum opus of sorts accumulating his lifelong work on solving the phase problem titled “Facing the phase problem”. This article describes how Dr. Hendrickson and his colleagues, including Dr. Smith, approached the phase problem, created solutions for it, and applied those solutions to modern technologies that now solve this problem with general ease. Dr. Smith then provides vital personal chronologies to the discussions in her follow up article in which she describes how Hendrickson, over the course of his career, systematically attacked this problem and developed the methodologies for which he is now famous. This biographical retelling of his work helps to humanize this great scientist and visualize his impact on the field of structural biology. The combination of Dr. Hendrickson’s scientific anthology and Dr. Smith's historical retelling reads together as a great biographical story of how great scientists are born and evolve into the giants we know today. Dr. Hendrickson's monograph and Dr. Smith's commentary can both be found in the International Union of Crystallography Journal.
Pictured below is PDB entry 1HR3, the first structure Dr. Janet L. Smith and Dr. Wayne Hendrickson published together in 1983 at the Naval Research Laboratory.
- Vida Storm Robertson, Fisk University
Vida Storm Robertson is a Masters Student in Chemistry at Fisk University working in both solid state and solution based structural determination techniques. He plans on starting a PhD program in biophysics next fall.
0 notes
Text
Radical Reactions - Yvain Nicolet - Institut de Biologie Structurale
Sharing some of our #SBGrid member tales from the last year. This one from January 2023.
The periodic table may be an icon of chemistry, but a cluster of elements at its center gives biology some essential pop and sparkle. Yvain Nicolet at the Institut de Biologie Structurale (IBS) in Grenoble, France, wants to learn exactly how those transition metals in the middle of the table put more pizzazz in proteins.
About one-third of proteins contain transition metals, such as cobalt, copper, molybdenum, or iron. The Nicolet lab mainly studies metalloproteins that contain iron-sulfur clusters but also scrutinizes enzymes that make these clusters. "Transition metals are often key components of the proteins in terms of function," Nicolet says. "They are only few atoms in a structure and sit in specific sites, but they enable specific activities, notably in enzymes, but not only in enzymes." Metals give proteins some superpowers in the form of new chemical properties and functions not otherwise possible with combinations of the 22 amino acids.
"They enhance the reactivity of the proteins by making many strange active sites that are very interesting," he says. These reactions can be blazingly fast with complex chemistry. And they are technically challenging to study.
The lab investigates enzymes that use metals, as well as proteins that build the metal cofactors and insert them into the enzymes. "We try to trigger the reaction in a crystal and to trap the different intermediates during the reaction to characterize by structural biology," Nicolet says.
Many metalloproteins evolved in a time before oxygen and can only make their razzle-dazzle moves in an anaerobic space. To prevent oxygen from destroying their molecules, experiments in the Nicolet lab take place in a series of six large anaerobic chambers filled with nitrogen.
The researchers reach in through gloves built into the airtight boxes to work with samples and equipment. There, cells are grown and broken up. The cell extracts are cleared and proteins separated by chromatography. Crystallization robots in one glove box screen for optimum conditions that researchers can manually reproduce in another box and freeze in liquid nitrogen. From there, the synchrotron is only 500 meters away.
"We determine 3D structures at moderate to high resolution," Nicolet says. "At first, we aim at determining where the metals are in a given protein, and how they interact with their environment (the protein matrix). We also aim at determining the structure of the metallocofactors or metal binding sites in terms of ligands and geometry.
"Then in a second step, we aim at understanding how these metals confer a specific activity to the protein," he continues. "For instance, in an enzyme, we would get intermediate states with the metal in a different coordinate and/or redox state to better understand what is going on during the reaction."
One milestone of the lab's work arose from successfully trapping an unexpected radical intermediate of NosL, a radical S-adenosyl-L-methionine (SAM) enzyme, in the process of generating a peptide with potent activity against gram-positive bacterial pathogens (Science, 2016).
Crystallography provided the position of the atoms, and for this paper they also used a spectroscopy technique called electron paramagnetic resonance (EPR), which gives the position of unpaired electrons near atoms.
Two months later, the group reported results from a study in which they carried out radical based chemistry catalyzed by the radical SAM enzyme HydE from beginning to end in a crystal (Nature Chemistry, 2016).
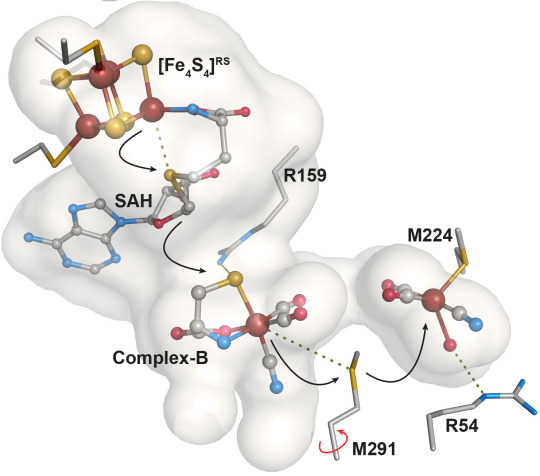
The image shows substrate processing and product transfer within HydE cavity during the FeFe-hydrogenase active site assembly process. Credit: Y.Nicolet
More recently, Nicolet's team characterized intermediates in the assembly of the nitrogenase, a key player in the global nitrogen cycle. The enzymatic complex nitrogenase catalyzes the reduction of nitrogen gas N2 to ammonia NH3, transforming nitrogen and returning it to plants. Specifically, the paper reported the X-ray structure of NifB protein from a nitrogen-fixing bacteria, one of a dozen proteins required to assemble the nitrogenase active site (Journal of the American Chemical Society, May 2020).
"For nitrogenase, there is a dedicated machinery with very, very challenging chemistry involved, and this is what we are attacking over the few next years," he says.
Nicolet was born and raised in Grenoble, France, where he now lives and works. Growing up in the city, he became an accomplished cello player with ambitions of becoming a professional musician. He started college in Grenoble. When a music school in Paris turned down his application, he chose to pursue science at what is now the University of Strasbourg. In Strasbourg, all the science classes emphasized the three-dimensional structures of molecules, whether it was enzymology, DNA replication, or proteins. He completed his undergraduate degree and stayed for graduate training in chemistry, biology, crystallography and nuclear magnetic resonance (NMR).
In a dinner discussion, a friend noted his interest in inorganic chemistry and suggested a structural biology lab in Grenoble working on metalloproteins. He moved back to complete his PhD at what is now the Université Grenoble Alpes. Nicolet worked on the structure of the [FeFe]-hydrogenase (an enzyme that makes or use molecular hydrogen), basic science with a potential application in producing alternative fuel.
From there, he joined the Drennan Lab at the Massachusetts Institute of Technology. In Cambridge, he discovered radical-based chemistry working on this newfound Radical SAM Proteins superfamily, now known for their ability to synthesize many vitamins, cofactors, or antibiotics.
He moved back to Grenoble for a one-year postdoctoral fellowship with the European Synchrotron Radiation Facility. In 2004, he joined IBS as a scientist and became a group leader there in 2016.
The Nicolet lab often dives into the molecular mechanisms of phenomenon discovered by other groups. "In my lab, we are not at the beginning of the discovery, and we are probably not at the end of the process," he says. "We focus on the molecular mechanisms inside the enzymes."
In a new challenge, the lab has set up a system to use cryo-electron microscopy to examine the assembly dynamics and reactions at room temperature without oxygen. Nicolet is interested in the basic science of radical chemistry, but he also hopes to be able to harness a radical enzyme and modify its catalytic activity to produce a molecule through directed evolution, such as a better antibiotic.
Nicolet also enjoys taking a bigger picture view of the nitrogen cycle, thanks to his garden. He came late to the activity and confesses that he is not good at growing vegetables. "You can observe the nature or the interaction between the different organisms that live together from bacteria to insects and plants," he says. "For me, it's the best way to contemplate what Darwin described as natural selection. And evolution. I'm not very good at producing vegetables, but I really enjoy seeing how all these organisms compete together or help each other as well.
-Carol Cruzan Morton
0 notes
Text
Second Takes - Andrea Thorn - University of Hamburg
Sharing some of our #SBGrid developer tales from the last year. This one from February 2023.
Andrea Thorn had been a junior group leader for a less than a year when, in early January 2020, researchers in China identified the cause of a mysterious contagious illness with pneumonia-like symptoms. Less than a week later, they released its genetic code, which revealed the novel virus to be a close cousin to the SARS coronavirus (SARS-CoV-1) that caused an outbreak in 2002-2003.

The rapid scientific response gave Thorn an idea for a talk she was due to give over coffee and cake to colleagues in her building at the Julius Maximilian University of Würzburg in Germany. She ran the only lab there focused on new computational methods for experimental structural biology. Most scientists in the building were infectious disease specialists.
"I needed to explain to them how my work was relevant," she says. Thorn works on ways to better model the atomic structures of RNA and proteins to make them useful for structure-based drug discovery, something that quickly would become more important as researchers rushed to respond to COVID-19.
Molecular models are a foundation of modern drug and vaccine development. They enable scientists to identify how to break the cycle of infection by targeting the right spot in the right protein. But even the most carefully determined structures reported in the most prestigious journals are imperfect interpretations of experimental data, Thorn says. Small errors can have large consequences in the search for a drug with the right fit.
When Thorn was preparing for her talk, no structures from the SARS-CoV-2 virus had been published yet. But more than 100 structures from SARS-CoV-1 were available in the worldwide Protein Data Bank (wwPDB). The structures ranged from a key protease in viral replication to the spike protein needed to attach and enter host cells.
From a random sample of five, Thorn found three SARS structures to illustrate the point of her research: Most protein structures can be improved with additional analysis.
Her talk was well received, germinating the seed of another idea that first seemed outlandishly bold and then became increasingly urgent as COVID-19 spread and the death toll soared. Thorn floated the idea over dinner with her mentor, Arwen Pearson in Hamburg: Analyze and correct all SARS structures. Pearson encouraged her, as did another senior colleague, Elspeth Garman of Oxford University.
In March 2020, as the first SARS-CoV-2 structures were being reported, Thorn first got her own small group on board and within a week pulled together a team of like-minded and mostly junior structural biology developers from all over the globe as the “Coronavirus Structural Task Force”. Many of them were international specialists in their respective fields, she notes.
They met online every weekday and posted daily updates to GitHub for anyone to access. Every Wednesday, when new wwPDB structures were released, their automated pipeline identified new coronavirus structures and assessed the quality of a representative sample of models and experimental data. When they improved a structure, they sent it back to the original authors to update the wwPDB entry, no strings attached.
For their web site (https://insidecorona.net), the group wrote blog posts to share with colleagues the larger story emerging from aggregated data of multiple structures. They wrote explanatory pieces for the public. They posted a 3D printable model of SARS-CoV-2.

Coronavirus Structural Task Force website: https://insidecorona.net. Image credit: Thomas Splettstößer
A year into the pandemic, structural biologists had released a total of 1,146 SARS structures covering 18 proteins from both SARS-CoV-1 and SARS-CoV-2. Most were derived by X-ray crystallography (73%) and single particle cryo-electron microscopy (cryo-EM) (24%) (Nature Structural & Molecular Biology, January 2021).
The task force's analytical pipeline deployed software programs from contributors inside and outside the Task Force. Among them were two tools previously developed by Thorn—AUSPEX, used for experimental X-ray data to detect sample measurement and processing problems (Acta Crystallographica, 2017) and, for experimental cryo-EM data, HARUSPEX, a neural network tool to automatically distinguish between nucleic acids and protein and to assign secondary protein structure elements (Angewandte Chemie, International Edition, 2020).
"We didn't do any peer reviewed publication until 2021," Thorn says about the task force. "We just wanted to fight the pandemic. The whole thing was fast. We pushed and pushed and pushed against corona so that drug developers and vaccine developers would have the right data. You know, it's been fantastic. It was born out of spontaneous willingness. I wish science would always be like this."
The task force published a description of its work (Nature Structural & Molecular Biology, May 2021). In 2023, Thorn says a dozen articles are in progress for peer-reviewed journals to summarize the combined information and results of their work, as well as to note questions yet to be answered about SARS-CoV-2. The task force will wind down in summer 2023. Thorn is strategizing on the next steps for a tenured academic position.
Thorn grew up in Germany in an intellectual family. Her father is a chemist, her mother an artist and entrepreneur. She is the third-born child with three brothers.
She calls her path to structural biology straightforward. One of her early science-related memories goes back to age 3, when her father explained surface tension to her with the gravity-defying demonstration of floating a needle on water. "My dad showed that to me with his always huge confidence that I would understand everything," Thorn says.
The family moved four times when Thorn was in elementary school. She supplemented the patchwork early education with natural curiosity. She read voraciously from the family library, made observations with a home microscope, and conducted chemistry experiments on the sly. In her mother's atelier, she also painted on large blank canvases.
In her teens, she and her friends were early adopters of live action role-playing games that have subsequently become popular in Germany. Participants assume the roles of characters and act out scenarios in a collaborative storytelling experience. To prepare for her roles, she researched scenarios ranging from futuristic military campaigns in Libya to glial cell retrieval from patients. "I read up to 600 pages a day when I was a teenager," she says.
In school, Thorn's studies became concentrated in chemistry and art. She continued first to a bachelor's degree and a master's degree, a prerequisite for her PhD.
She excelled in the lab and in computational chemistry, but her breakthrough lessons came after she failed quantum chemistry. She sought a tutor in the solid-state physics department, where her academic track record was unknown. A professor she consulted for a referral, Helmuth Zimmermann, volunteered to oversee weekly tutoring for Thorn and other classmates who needed help.
"Arguably those tutoring sessions did more for my education than everything else that semester," she says. "I'd never used maths before to describe things in nature in that way and fell in love with that."
One day, in response to Thorn's curiosity, the professor explained his research, which was measuring crystals to determine the structure of molecules. "I was hooked, because that meant the maths that I just learned could be applied far beyond class," she says. "It meant you could use them to find out the structure of crystal lattices and, with the structure of crystal lattices, the structure of molecules."
She skipped her prescribed medicinal chemistry classes in favor of crystallography lectures, which were not in her curriculum plan. One day, her crystallography lecturer refused to answer her question about SHELX, a crystal structure refinement program, saying she wasn't smart enough to understand.
"I was so angry, I printed out the SHELX manual, which is 100 pages, and I read the whole fricking thing." She came to the next class armed with new and more detailed questions. "It turned out he didn't know so much about crystallography," she says.
When it came time to choose a PhD group, Thorn had a list. The list included George Sheldrick at University of Göttingen, who developed SHELX. On the way to a holiday live role-play destination, she stopped by his lab to scout it out informally. Unexpectedly, she had a long conversation with him that ended with an immediate offer of doing her PhD thesis in his group.
She thrived in the high-performing lab and found fellow role-play enthusiasts. When she was teaching her first classes as a PhD student, a friend came to collect her for lunch. He noted her stance in front of the students (who were much older than her) looked like the last weekend's live role play when she had played a commanding leader with feet planted and shoulders squared as she led her warriors into battle.
In 2011, Thorn graduated with her doctorate and four new first-author publications from her thesis. The morning after her defense, she married her childhood sweetheart, a chemist. Within a year, she had secured a prestigious three-year Marie Curie fellowship for independent research at the MRC Laboratory of Molecular Biology in Cambridge, followed by a brief but productive stint at University of Oxford.
Homesick for Germany and their parents, she returned with high expectations of becoming an assistant professor, something that remains a goal. She secured outside funding and became a group leader at University of Würzburg in 2019 and moved to Universität Hamburg in 2020.
The task force may be winding down, but its value lingers in Thorn's mind as an unmet need in science. She has been thinking about the volume of data being generated by the structural biology community and how to combine the data for more meaningful interpretation and usefulness. She also takes it as an example of what is possible when experts across the globe start to collaborate on an important problem.
"We are generating data like crazy," she says, “beyond the capacity of individuals to assimilate and make sense of details from different techniques across labs. "In Germany, we have this beautiful word, Datennachnutzung. It means you are using experimental data for a second time to find new insights. And that's something we absolutely need to do."
-Carol Cruzan Morton
1 note
·
View note
Text
How one amino acid can affect light absorption of bacteria
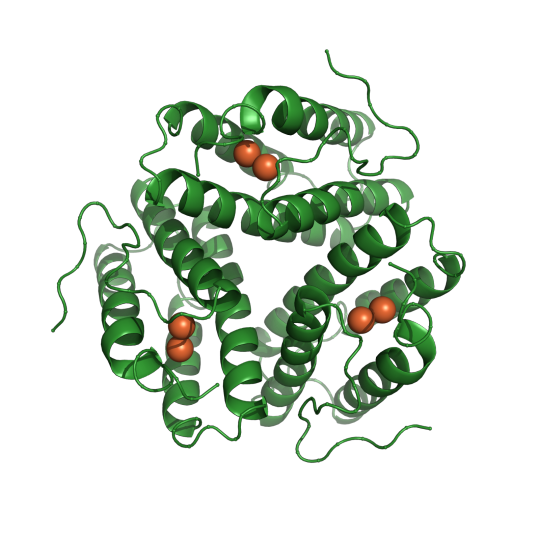
Plants and bacteria use light for energy and to carry out many essential functions, but scientists are still trying to understand how they are able to harness light from the sun. Proteins can form unique shapes and arrangements that allow them to somehow transform light into energy and signals, but the mechanism for such actions is not clearly understood. One such protein is found in cyanobacteria and is used to help these bacteria make decisions about movement and aggregation based on the quantity of light they receive. The protein responsible is named cyanobacteriochrome Slr1393 and in a study published in the Journal of Molecular Biology, SBGrid member Katrina Forest and colleagues investigate the interesting ability of this protein to convert from absorbing red light to absorbing green light.

Pictured above is Cyanobacteriochrome Slr1393 (teal). Tryptophan 496 is shown in purple overlapping with PCB chromophore (green). PDB:5DFX. CC BY SBGRID
Using a large toolkit of different spectroscopies, including UV-vis, IR and UV-RR, this team of researchers showed that the switching abilities of this protein appear to be caused by the alignment of a singular tryptophan residue. In the red absorbance confirmation it appears that the tryptophan residue is stacking with other cyclic and conjugated molecules. Spectroscopy results suggest that this double bond stacking helps this protein absorb light in the red wavelengths. When the tryptophan residue is moved out of a stacking arrangement, spectra results suggest that the protein is more adept at absorbing light in the green wavelengths. This theory was supported with mutant proteins that replace this key tryptophan with a less optically active residue, valine. Spectra showed that this valine mutated protein absorption did not have much meaningful change when converting between red and green absorbing conformation. These results suggest that the absorption properties of light sensitive proteins can be very sensitive and reliant on the precise 3D structure of these molecules.
Read more in the Journal of Molecular Biology.
-Vida Storm Robertson, Fisk University
1 note
·
View note
Text
All-in-one shock and kill therapies could be the future of HIV-1 treatment
Human immunodeficiency virus type 1, or HIV-1, is the most common type of HIV and attacks a person’s immune system, preventing them from fighting off infections. Although there is currently no cure for HIV, cART (combination antiretroviral therapy) is very effective at treating HIV and reducing the viral load to undetectable levels. However, most people have to continue cART for the rest of their lives due to the phenomenon known as viral rebound, when persistent detectable levels of viral particles come back after treatment is discontinued. The need for lifelong cART presents many issues and is why scientists stress the need for a cure for HIV-1. One approach to a cure that is gaining in popularity is the shock and kill method. This method involves reactivating the latent HIV-1 proviruses (an inactive form of the virus that has integrated into the host DNA) and then killing the virus-producing cells. SBGrid member Andreas Plückthun and colleagues expanded on this method to create a novel all-in-one shock and kill therapy to treat HIV-1.
In this work, Plückthun and colleagues used a CRISPR activation system (the shock) with a truncated form of the human pro-apoptotic protein Bid to create a suicide gene (the kill), and combined both into an adenoviral vector that was repurposed to inhibit natural tropism and for use in different cell types. These components combined created their novel all-in-one therapeutic strategy. Their work showed that this therapy is able to eliminate infected cells in an HIV-1 and T cell specific manner before they release particles to further the spread of infection. This method is also highly flexible and customizable to meet the needs of individuals with HIV-1.
Read more about this work in Gene Therapy.
- KeAndreya Morrison, Meharry Medical College
1 note
·
View note