#trinucleotide repeat
Text
Future Child Diagnosed with Huntington's Disease
Hi everyone, I am 4.5 months pregnant and just got a pre-natal genetic test done which revealed that the child I am carrying will be diagnosed with Huntington's disease. I don't completely understand everything just yet, so if anyone with similar experiences has any advice, I'd really appreciate it! Here is what my doctor said and what my own research has amounted to so far:
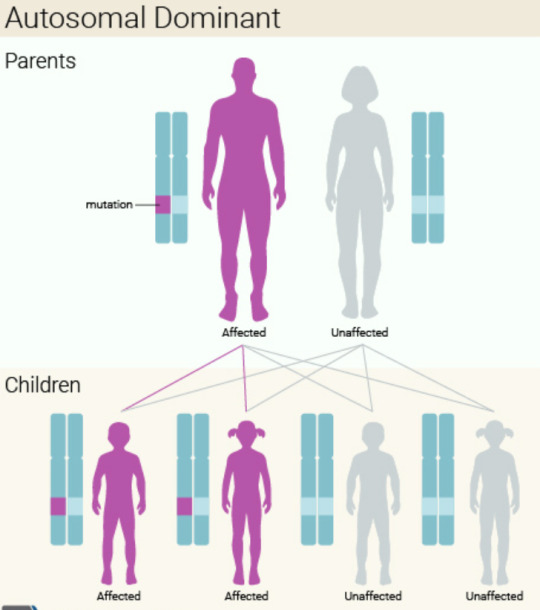
How my child inherited it unknowingly: Huntington’s disease is an autosomal dominant disorder, which means that a person only needs one copy of the gene to develop the disorder. It is caused by mutations in the HTT gene. The HTT gene provides the instructions for making a protein called huntingtin, too much of the protein is toxic to nerve cells. The HTT gene has a DNA segment known as the CAG trinucleotide repeat which is usually repeated 10 to 35 times. In people with Huntington’s disease, the segment repeats 36 to more than 120 times, however, those whose segment repeats 36 to 39 times may not develop symptoms. My husband has the gene and his CAG segment repeats 37 times, but he thankfully won’t develop symptoms as he’s already reached the general age of development, his 30s or 40s, however, he has passed down the gene to our future child.
Symptoms:
Causes the degeneration of nerve cells in the brain.
Affects functional abilities and usually results in movement, cognitive, and psychiatric disorders.
Movement disorders: chorea (involuntary jerking or writhing movements), dystonia (muscle problems such as rigidity or muscle contracture), slow or unusual eye movements, impaired walking, posture, and balance, and difficulty with speech or swallowing.
Cognitive disorders: difficulty prioritizing or focusing on tasks, tendency to get stuck on a thought, behavior, or action, lack of impulse control, lack of self-awareness, slowness in processing, and learning difficulties.
Psychiatric disorders: feelings of irritability, sadness, or apathy, social withdrawal, insomnia, fatigue, suicidal thoughts. OCD, mania, or bipolar disorder.
Causes of death: pneumonia or other infections, injuries related to falls, complications related to the inability to swallow.
Treatment: The disease is unfortunately not curable, but medications are available to help manage the symptoms. Medications include drugs for movement control such as tetrabenazine and deutetrabenazine, but they may cause drowsiness, restlessness, and the risk of worsening or causing depression or other psychiatric conditions. Antipsychotic drugs such as haloperidol and fluphenazine may be beneficial in treating chorea but may worsen dystonia, restlessness, and drowsiness. Quetiapine and olanzapine may help suppress violent outbursts and other mood disorders, however, may cause movement disorders themselves. Antidepressants such as citalopram, escitalopram, fluoxetine, and sertraline may also treat OCD, however, may cause nausea, diarrhea, drowsiness, and low blood pressure. There is also psychotherapy, speech therapy, and physical therapy.
There also seems to be some current research being done by scientists. They are trying to understand the toxicity of the mutant huntingtin protein and how to develop a potential drug to counteract it. Scientists are using cutting-edge methods (hope it works!!) such as optogenetics (neurons activated or silenced in animal brains using light beams) to study circuit defects in Huntington’s disease and are using stem cells to study disease mechanisms and to test potential therapeutic drugs. My child will have many years until the symptoms start so I hope that the research will become tangible enough to relieve them of their future symptoms.
This is all really just scientific research so if any other parents in the community have any advice, please post it!!
5 notes
·
View notes
Text
Imperfect hairpins formed by CTG trinucleotide repeats are heteroduplex DNA molecules in living cells
BioRxiv: http://dlvr.it/T7rkr9
0 notes
Quote
Expansions of CAG trinucleotide repeats cause several rare neurodegenerative diseases. The disease-causing repeats are translated in multiple reading frames and without an identifiable initiation codon. The molecular mechanism of this repeat-associated non-AUG (RAN) translation is not known. We find that expanded CAG repeats create new splice acceptor sites. Splicing of proximal donors to the repeats produces unexpected repeat-containing transcripts. Upon splicing, depending on the sequences surrounding the donor, CAG repeats may become embedded in AUG-initiated open reading frames. Canonical AUG-initiated translation of these aberrant RNAs may account for proteins that have been attributed to RAN translation. Disruption of the relevant splice donors or the in-frame AUG initiation codons is sufficient to abrogate RAN translation. Our findings provide a molecular explanation for the abnormal translation products observed in CAG trinucleotide repeat expansion disorders and add to the repertoire of mechanisms by which repeat expansion mutations disrupt cellular functions.
CAG repeat expansions create splicing acceptor sites and produce aberrant repeat-containing RNAs: Molecular Cell
0 notes
Text
Metabolomics: An Emerging "Omics" Platform for Systems Biology and Its Implications for Huntington Disease Research
Huntington's disease (HD) is a progressive, fatal neurodegenerative disease characterized by motor, cognitive, and psychiatric symptoms. The precise mechanisms of HD progression are poorly understood; however, it is known that there is an expansion of the trinucleotide cytosine-adenine-guanine (CAG) repeat in the Huntingtin gene. Important new strategies are of paramount importance to identify early biomarkers with predictive value for intervening in disease progression at a stage when cellular... http://dlvr.it/T0Ykcm
0 notes
Text
Abstract
To obtain information of Periplaneta americana, we analyzed the distribution characteristics of microsatellite sequences in the P. americana transcriptome (229 MB) by using MSDBv2.4. The total number of perfect microsatellite sequences was 38 082 and covered about 0.3% of P. americana transcriptome. The cumulative length of microsatellites was 618 138 bp, and the density of microsatellites was 2978.54 bp/Mb. In the different repeat types of the microsatellites, the number of the mononucleotide repeats was 20 002 (accounting for 52.52%), which obviously was the most abundant type. While the trinucleotide, tetranucleotide, dinucleotide, pentanucleotide and hexanucleotide repeats accounted for 24.51, 12.97, 8.13, 1.61 and 0.26%, respectively. The kind of different repeat copy categories in each repeat type was also quite different, such as the A in mononucleotide repeat type, the AG in dinucleotide, the AAT in trinucleotide, AAAT in tetranucleotide, the AAGAA in pentanucleotide, and the CAGTAG in hexanucleotide were the most of each category. The A, T, AC, AG, AT, GT, AAG, AAT, ATC, ATG, ATT, CTT, AAAG and AAAT were the dominant repeat copy categories, the total number of all these types was 29 933, accounting for 78.6% in the total number of microsatellite sequences. These results based on a foundation for developing high polymorphic microsatellites to research the functional genomics, population genetic structure and genetic diversity of P. americana.
0 notes
Text
Dysregulated #miRNA and #mRNA Expression Affect Overlapping Pathways in a Huntington's Disease Model
Huntington's disease (HD) is a fatal neurodegenerative disorder caused by the expansion of a CAG trinucleotide repeat in the Huntingtin gene. Transcriptional dysregulation is one of the main cellular processes affected by mutant Huntingtin (mHtt). In this study, we investigate the alterations in #miRNA and #mRNA expression levels in a Drosophila model of HD by #RNA sequencing and assess the functional effects of misregulated #miRNAs in vivo. We found that in head samples of HD flies, the level of 32... https://pubmed.ncbi.nlm.nih.gov/37569316/?utm_source=dlvr.it&utm_medium=tumblr&utm_campaign=None&utm_content=1Zap-74u4XbiV7x0qz5lToBuxtoq00qwwHZUuXSRQOsim8UYds&fc=None&ff=20230816100626&v=2.17.9.post6%2086293ac
0 notes
Text
Fragile X Syndrome: Unraveling the Genetics and Characteristics
Understanding Fragile X Syndrome: The FErTILe X Syndrome
Introduction:
Fragile X syndrome (FXS) is a well-known genetic disorder characterized by the expansion of the trinucleotide repeat CGG in the fragile X mental retardation 1 (FMR1) gene. This condition is often associated with specific physical and cognitive characteristics, affecting both males and females. In this article, we will explore…

View On WordPress
0 notes
Text
What is the association between molecular variations in the fragile X gene and fragile X syndrome?
Fragile X syndrome is caused by a mutation in the FMR1 (fragile X mental retardation 1) gene, which is located on the X chromosome. Specifically, individuals with fragile X syndrome have an expansion of a trinucleotide repeat sequence (CGG) in the FMR1 gene. This expansion leads to a loss of function of the FMR1 protein, which is important for normal brain development and function. The number of…

View On WordPress
0 notes
Photo
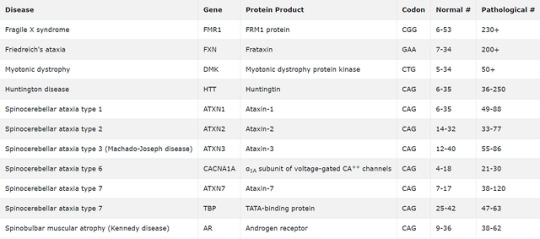
Huntington disease is characterized by choreoathetoid movements of the face and extremities, progressive dementia, and early death. Huntington disease has autosomal dominant inheritance, not X-linked dominance or autosomal recessive inheritance. Huntington disease is one of the trinucleotide repeat disorders (others include a number of the spinocerebellar ataxia diseases and Fragile X syndrome) and is due to a mutation on chromosome 4 that results in excessive nucleotide CAG repeats, which result in a longer than normal string of glutamine residues (polyQ expansion) in the N-terminal region of the protein huntingtin. Huntingtin plays a role in vesicle and organelle transport along axons, and increasing the length of the polyQ region disrupts this function. In addition, polyQ expansion has also been implicated in disrupting transcription of a number of genes. Expansion of these repeats over successive generations leads to earlier manifestations of the disease in affected offspring, a phenomenon known as anticipation. Above is a table of the common trinucleotide repeat disorders. The table misspelled "Friedreich!" "I" before "e!"
In Friedreich ataxia, you get inability to regulate iron-> free radical damage.
Bottom Line: Huntington disease is an autosomal dominant disease caused by an inherited mutation in chromosome 4 in which trinucleotide repeats result in impaired axonal transport and decreased and ineffective transcription of a number of genes.
I remember in B&B, he said that glutamate binding to NMDA receptor-> excessive Ca2+ influxe-> neuronal death.
From Medscape:
The most striking neuropathology in HD occurs within the neostriatum, in which gross atrophy of the caudate nucleus and putamen is accompanied by selective neuronal loss and astrogliosis. Marked neuronal loss also is seen in deep layers of the cerebral cortex. Other regions, including the globus pallidus, thalamus, subthalamic nucleus, substantia nigra, and cerebellum, show varying degrees of atrophy depending on the pathologic grade.
The extent of gross striatal pathology, neuronal loss, and gliosis provides a basis for grading the severity of HD pathology (grades 0-4).
No gross striatal atrophy is observed in grades 0 and 1. Grade 0 cases have no detectable histologic neuropathology in the presence of a typical clinical picture and positive family history suggesting HD. Grade 1 cases have neuropathologic changes that can be detected microscopically but without gross atrophy. In grade 2, striatal atrophy is present, but the caudate nucleus remains convex. In grade 3, striatal atrophy is more severe, and the caudate nucleus is flat. In grade 4, striatal atrophy is most severe, and the medial surface of the caudate nucleus is concave.
The genetic basis of HD is the expansion of a cysteine-adenosine-guanine (CAG) repeat encoding a polyglutamine tract in the N-terminus of the protein product called huntingtin.
The function of huntingtin is not known. Normally, it is located in the cytoplasm. The association of huntingtin with the cytoplasmic surface of a variety of organelles, including transport vesicles, synaptic vesicles, microtubules, and mitochondria, raises the possibility of the occurrence of normal cellular interactions that might be relevant to neurodegeneration.
N-terminal fragments of mutant huntingtin accumulate and form inclusions in the cell nucleus in the brains of patients with HD, as well as in various animal and cell models of HD.
The presence of neuronal intranuclear inclusions (NIIs) initially led to the view that they are toxic and, hence, pathogenic. More recent data from striatal neuronal cultures transfected with mutant huntingtin and transgenic mice carrying the spinocerebellar ataxia-1 (SCA-1) gene (another CAG repeat disorder) suggest that NIIs may not be necessary or sufficient to cause neuronal cell death, but translocation into the nucleus is sufficient to cause neuronal cell death. Caspase inhibition in clonal striatal cells showed no correlation between the reduction of aggregates in the cells and increased survival.
Furthermore, postmortem studies reveal that NIIs are quite rare in the striata of patients with HD as compared to the cortex, and most of the aggregates within the striatum are observed in populations of interneurons that typically are spared in individuals with HD.
#Huntingtons disease#Huntington#Huntingtons#dementia#neuro#fragile X syndrome#friedrichs ataxia#friedrich ataxia#freidrich#myotonic dystrophy#spinocerebellar ataxia#Kennedy disease#spinobulbar muscular atrophy#genetics#trinucleotide repeat#glutamate
1 note
·
View note
Text
molecular mechanisms of mutation, DNA repair, and allelic fixation
mutation event may or may not result in a heritable mutation via following process:
initial alteration: DNA damage generates some altered sequence
depurination - A or G hydrolyzes and drops off DNA sugar-phosphate backbone, leaving a blank apurinic site in a template parent strand
oxidative deamination - C loses its NH3 group and becomes U, so that template parent strand encodes A-T base pair instead of C-G
radiation - X-rays cause a double-strand break and an entire portion of the helix code lost
thymine dimers - bond created between two T’s on the same strand via ring-conformation-changing UV light
oxidation - O eliminates across G’s C=N double bond and forms 8-oxodG, which pairs with A instead of C (transversion)
mutagen - physical or chemical agent that causes mutation rate above natural spontaneous levels
base analog mutagen - similar form to bases but different pairing
hydroxylating agent - adds OH group to base
alkylating agent - adds CH3 or CH2CH3 group to base
deaminating agent - removes NH3 group from base
intercalator - particulate insertion between base pairs and subsequent disruption of DNA replication machinery, increasing single nucleotide insertion/deletion events
initial alteration: DNA replication machinery mistake generates some altered sequence
DNAP makes a mistake - very rare, ameliorated even more so by 3′ - 5′ exonuclease subunit
base tautomerization - base on parent strand is in rare tautomeric form, which pairs differently than usual base; one daughter helix receives incorrect tautomeric pairing
slipped mis-pairing - pausing DNAP allows for either template or daughter strand to slip from its grasp, and wrongly placed pairing occurs
pre-mutation trinucleotide repeats - large number of repeats that are not quite over phenotypic threshold quantity, but replication machinery is likely to slip and make more/less repeats in next DNA replication process
DNA repair: mechanisms for complete reversion to WT sequence
DNA repair mechanisms may catch potential mutations before they become fixed in the next replication round
alkyltransferase - removes miscellaneous methyl and ethyl groups added randomly
photolyase - removes thymine dimers in the presence of visible light via chromophore photorepair
DNA glycosylase - removes certain incorrect bases from sugar-phosphate backbone and nicks it in base excision repair, so that AP endonuclease and phosphodiesterase clear surrounding nucleotides/backbone and DNAP re-synthesizes DNA portion
UvrA/B/C complexes - remove incorrect bases for which base-specific DNA glycosylases do not exist in nucleotide excision repair, scanning for helical irregularities and cutting out damaged portions for later DNAP re-synthesis
adenine methylase - places methyls on template parent strands during replication rounds for detection by post-replication proteins MutL/S/H in methyl-directed mismatch repair, which nick the un-methylated, misspaired daughter strand for DNAP re-synthesis | MutL recognizes CH3, MutH nicks unmethylated strand
DNA repair: mechanisms for possible reversion to WT sequence
DNA repair mechanisms for double-strand breaks may or may not result in reversion to WT depending on repair method, cell cycle stage, and individual’s genotype
homologous recombination - one sister chromatid experiences DNA loss via double-strand break
DNAP utilizes either other sister chromatid during mitosis or meiosis, or homolog chromatid during interphase, as template for repair
if homolog is template, new DNA may or may not be same as original allelic sequence depending on homo- or heterozygotism at relevant locus
non-homologous end-joining (NHEJ) - occurs in G1 without sister chromatids for recombination
broken-off segment is bound to at ends by protein complexes to keep from degrading; rejoined to original via DNA ligase
genes or informant sequence portions at ends of breaks may denature before NHEJ protein complexes can bind and preserve them - results in loss of genetic material and possible phenotypic alteration
micro-homology-mediated end-joining (MMEJ) - occurs after failure to repair double-strand break via either homologous recombination or NHEJ
3′ ends of double-strand breaks are exposed to help guide broken ends back to origins
purposeful exposure-intended DNA cutting, like NHEJ denaturing before protein complex binding, results in small deletions at ends of break, although majority of sequence is still intact
SOS system - error-prone “sloppy” DNAPs repair severely damaged DNA by throwing random nucleotides into place of broken segments
fixed mutation: occurs after non-repair and second-round replication of altered sequence
DNA damage or replication mistake is not caught before new replication cycle
altered sequence is passed down to 1 of 2 daughter strands - after first F1 replication round, mutations can still be detected
without DNA repair, second F2 replication round produces fully fixed altered sequence in the genome that is now a genetic mutation in the mutated daughter strand
macroscopic considerations for mutation mechanisms
mutations in DNA repair machinery result in high risk for genetically-based diseases due to dysfunctional DNA repair after environmental damage
species-wise, larger allelic frequencies rest with mutations/DNA sequences that encode for beneficial phenotype
13 notes
·
View notes
Photo

Genetics
Secondary to a mutation on the short arm of chromosome 4, in the HTT (aka IT15) gene: a CAG trinucleotide repeat, which encodes for the huntingtin protein.
HD is an autosomal dominant disorder with significant genetic anticipation: the sequence length is unstable and can expand during meiosis, especially down paternal inheritance lines.
It encodes a polyglutamine ('polyQ') stretch at the N-terminus of the huntingtin protein.
The anticipation helps us remember that this is a trinucleotide repeat disorder, specifically a CAG repeat disorder:
Classic trinucleotide repeat expansion disorders
Huntington's disease (CAG), myotonic dystrophy (CTG), and fragile X syndrome CGG) are the three classic trinucleotide repeat expansion disorders: the trinucleotide sequence is repeated many times in a row.
Additional CAG repeat disorders include: Kennedy's disease (aka X-linked spinal and bulbar muscular atrophy), spinocerebellar ataxia type 1 (SCA 1) and type 3 (SCA3, aka Machado-Joseph disease).
Pathology ("CAG" mneomnic)
We use the acronym CAG to highlight some key aspects of HD neuropathology:
On gross examination, there is caudate (C) and putamen (aka striatum) atrophy (A) with resultant anterior horn dilatation.
Accordingly, there is a loss of striatal GABAergic (G) medium spiny neurons, which is what primarily constitutes the striatum.
Mneomnic via Number 4
The HD gene is on chromosome 4
Greater than 40 CAG repeats is abnormal (but, truly, anything more than 36 can be symptomatic)
The average age of onset is 40 years-old but the longer the repeat length, the earlier the age of onset.
Onset younger than 20 years old, is referred to as Juvenile HD; it manifests with an akinetic-rigid syndrome, rather than chorea, referred to as the Westphal variant, which is typically the end-stage of HD in adults.
Clinical
HD is predominantly a neuropsychiatric and movement disorder.
It's often mistaken as alcoholism early on but ultimately becomes parkinsonian, later.
Typical survival from onset is 15 years, much like the timeline of degeneration in Parkinson's disease.
Key domains:
Psychiatric. Depression and anxiety, early, and obsessive/compulsive thoughts, profound apathy, and physical aggression, later. Note that suicide is the 2nd most common cause of death in HD.
Cognitive. Executive dysfunction, early, such as trouble with organizational tasks, planning, and task sequencing.
Simple abnormal involuntary movements: tics, dystonia, myoclonus.
Complex abnormal involuntary movements: Chorea (excessive movements that flow from body part to body part).
Failure of voluntary movements: Akinetic, rigid syndrome (parkinsonism).
#ditki#pathology#chorea#huntington's disease#huntingtondisease#medstudyblr#medstudy#medicalstudent#medicalschool
12 notes
·
View notes
Text
What Is Huntington's Condition

Huntington's illness (HD) is an inherited condition impacting the mind function in a modern manner. This implies that it can be sent from parents to children which its onset might begin fairly unobserved, with slow-moving as well as stable growth influencing the individual.
Many frequently, Huntington's signs appear in grownups aged 35-44. If it develops before the age of 20, it is identified as adolescent Huntington's disease. An early-onset indicates slightly different symptoms as well as can advance faster than the common HD.
Individuals impacted by Huntington's condition generally survive on for concerning 15-18 years after the onset, in the case of the typical type of the disease, and also regarding 10-15 years for the juvenile HD. Nevertheless, sometimes signs are not present until their 50s or 70s.
For European populations, the ordinary occurrence of Huntington's condition is around 9-17:100,000. The condition is less common in other populations, with worths varying from 0.1 to 2 in 100,000 within indigenous African populaces in South Africa, as an example.
What creates Huntington's condition?
Huntington's disease is triggered by mutations in the HTT genetics. This gene offers directions for making huntingtin, a healthy protein that shows up to play a crucial function in nerve cells in the brain. It was the initial disease-associated gene to be molecularly mapped to a human chromosome in 1983.
Particularly, the anomaly that creates Huntington's condition is a CAG trinucleotide repeat i.e., a series of cytosine, adenine and also guanine that is repeated numerous times. As a whole, we anticipate to see 10-35 repetitions of these trinucleotides in the HTT genetics. Nonetheless, in individuals with Huntington's condition, the CAG sector is duplicated 36 to 120 times or even more.
The CAG repeat dimension will certainly figure out the phenotype:
-- 26 or fewer CAG repeats: typical
-- 27-35 repeats: there is no danger of creating the signs of the condition, nevertheless, it is feasible that their kids will create the condition because of instability of the CAG trinucleotide that may boost in the next generations.
-- 36 or even more repeats: these individuals are considered to have a lifetime threat of creating Huntington condition, however they are also classified into:
o 36-39 repeats: thought about to have a reduced penetrance allele. This suggests that the individuals go to risk of establishing the illness but might not create signs and symptoms of the illness, it is common that with this number of repeats people are asymptomatic.
o 40 or even more repeats: they are taken into consideration to have a full penetrance allele. That is, all persons having this number of repeats will certainly establish signs of the disease It has been revealed that the higher the number of trinucleotide repeats, the a lot more severe and larger the range of symptoms such as motor, cognitive and also functional disability can be.
There is a connection in between the number of repeats and the age of onset: 36-55 repeats usually appear in the grown-up kind, while over 60 repeats are present in people with the adolescent kind of the disease.
The modification in the genetics, creates an unusually long huntingtin healthy protein, which is cut into smaller, harmful pieces that bind together, translocate within the core, changes gene transcription, mitochondrial function, interrupting their regular feature, leading to cell death. The dysfunction and, at some point, death of these nerve cells in particular parts of the brain result in the onset of symptoms of Huntington's disease.
Signs and symptoms of Huntington's condition.
How can Huntington's condition be determined and also what symptoms should signal to this problem? Bearing in mind that symptoms come to be a lot more severe as the condition proceeds, we can split them right into groups based upon the timeline of their occurrence.
Signs and symptoms in the early stages
● Clumsiness
● Lethargy
● Irritation
● Stress and anxiety
● Disinhibition
● Delusions
● Clinical depression
● Olfactory disorder
Mid-disease Huntington's signs
● Dystonia
● Chorea, turning motions, jerking, incredible, unconnected stride
● Problems with activities calling for manual dexterity
● Slow voluntary activities; problem launching motion
● General weakness
● Weight loss
● Speech difficulties
● Stubbornness
End of illness signs and symptoms:
● Strength
● Bradykinesia
● Serious chorea (much less common).
● Inability to walk and/or talk.
● Trouble ingesting, which may be a choking risk.
● Failure to look after themselves.
Typical psychological conditions associated with Huntington's disease consist of:.
Obsessive-compulsive condition-- with intrusive ideas and also repetitive habits.
Mania-- with elevated state of mind, overactivity, impulse habits as well as pumped up self-esteem.
Bipolar illness-- with alternating episodes of anxiety as well as mania.
The majority of these are likewise existing face to faces with adolescent Huntington's disease, such as movement, mental and also psychological issues. Furthermore, juvenile affected persons will certainly experience slow motions, clumsiness, constant falls, tightness, slurred speech as well as salivating.
Huntington illness therapy alternatives.
While there is no well-known cure for Huntington's condition, persons influenced can obtain therapy specific to their symptoms as they show up.
For instance, for chorea movements, medicines might be prescribed, and anti-parkinsonian agents are additionally effective in enhancing conditions of hypokinesia and rigidness. For those experiencing psychotic signs, antidepressants can prove effective. Depending upon each client, there are a range of drugs that can act on certain signs. Professionals will recommend on these on a case-by-case basis.
Psychotherapeutic assessments can be extremely useful once the illness has been identified, given that they can resolve several symptoms including behaviour issues. With a physician, people can create future coping methods and also they can also manage assumptions of the condition for the patient and their family.
Another crucial area for therapy in Huntington's illness is managing difficulties with the muscular tissues required for ingesting.
Lastly, physical treatment can function to find out workouts establishing toughness and adaptability, along with control and also balance in affected persons. This will certainly help them keep movement as well as an independent way of life for as long as possible while dealing with the condition.
Inheritance of Huntington's condition.
Huntington's condition is acquired in an autosomal leading manner, suggesting that if only one duplicate of the gene is modified, the disease will appear. Therefore, an individual influenced by Huntington's illness has a 50% chance of transmitting it to their children. Research studies have actually shown that individuals with both copies of the genetics affected have a comparable age of start than people with simply one copy however may reveal an increased rate of condition progression.
When the altered HTT gene is handed down across generations the variety of these repeats increases. This causes a rise in condition intensity and/or decrease in age of beginning in successive generations-- a sensation referred to as anticipation. The growth of the CAG trinucleotide in the HTT genetics can be determined through genetic testing.
BGI China has a professional clinical team of hereditary counsellors that can aid comprehend your danger for certain genetic diseases. Do not hesitate to contact us in case you require more info.
Click to know more about BGI gene test products if you are interested.
0 notes
Text
Trey Gray-A Champion for Huntington's Disease
Trey Gray-A Champion for Huntington’s Disease
May is Huntington’s Disease Awareness Month!!
I love Trey Gray and I consider him a Champion for Huntington’s disease (HD) like Michael J. Fox is for Parkinson’s disease.
HD is an autosomal dominant trinucleotide repeat disorder that causes the progressive degeneration of the basal nuclei. This degeneration leads to clinical symptoms affecting voluntary movement, cognitive impairment, and…

View On WordPress
0 notes
Text
Retrotransposon expression is repressed in Huntington's disease
Huntington's disease (HD) is a dominantly inherited neurodegenerative disease characterized by expansion of the number of trinucleotide CAG repeats in the first exon of the HTT gene encoding the protein huntingtin. While the normal number of these repeats varies between six and 35, pathology is observed when the number gets above that, maxing out at around 150 repeats.
Source Link Retrotransposon expression is repressed in Huntington's disease
0 notes
Text
Functional Characterisation of the Circular #RNA, circHTT(2-6), in Huntington's Disease
Trinucleotide repeat disorders comprise ~20 severe, inherited, human neuromuscular and neurodegenerative disorders, which result from an abnormal expansion of repetitive sequences in the DNA. The most common of these, Huntington's disease (HD), results from expansion of the CAG repeat region in exon 1 of the HTT gene via an unknown mechanism. Since non-coding #RNAs have been implicated in the initiation and progression of many diseases, herein we focused on a circular #RNA (circ#RNA) molecule... https://pubmed.ncbi.nlm.nih.gov/37174737/?utm_source=dlvr.it&utm_medium=tumblr&utm_campaign=None&utm_content=1RYYbE7j9SUSBe_aHniaI_J1MQIFIBbfLuFxoWdLNMNDzVVIWF&fc=None&ff=20230516100620&v=2.17.9.post6%2086293ac
0 notes
Text
Fwd: Graduate position: UOslo.Genomics
Begin forwarded message: > From: [email protected] > Subject: Graduate position: UOslo.Genomics > Date: 16 March 2016 at 05:13:35 GMT > To: [email protected] > > > > > See below: > > Department of Biosciences, University of Oslo > PhD Research Fellowship in Genomics > Position as PhD Research fellow available at the Department of Biosciences. > > Online: > > > https://ift.tt/2X2PsIa > > > The fellowship will be for a period of 3 years, with no compulsory > work or for a period of 4 years, with 25 % compulsory work (teaching > responsibilities at the department) contingent on the qualifications of > the candidate and the teaching needs of the department. Starting date > no later than 01.10.2016. > > No one can be appointed for more than one fixed-term period at the > same institution. > > Job/ project description: > > The PhD fellow will be appointed on the project entitled "Evolutionary > and functional importance of simple repeats in the genome" funded by > the Research council of Norway through the FRIPRO program and lead > by Professor Kjetill S Jakobsen. The candidate will be working in > an internationally leading research group recently awarded Frontier > Research status. > > The project focuses on the functional modulation of regulatory > mechanisms affecting the phenotype by variations in simple trinucleotide > repeats residing inside (coding) and in the vicinity (or in introns) > of genes. Specifically, we would like to test the hypothesis that > hypervariable coding/regulatory repeats are promoting the ability of a > species or population to adapt to a changing environment. To address > this we will use Atlantic cod and Arabidopsis as model systems. The > project is cross-disciplinary and will utilize genomic, bioinformatics, > statistics and experimental approaches and the appointed PhD candidate > will work in a larger team and in close connection with an additional > PhD that will be hired concurrent with this position. > > The PhD candidate will investigate the genomic architecture of > repeats and repeat-length variation in individuals of Atlantic cod and > Arabidopsis using PacBio long read technology. A particular focus will > be on simple repeats within or in the vicinity of genes. Data from > the Aqua Genome Project and the 1001 Arabidopsis genome project will > also be investigated. Further, trans-generational variation in repeat > length will be addressed. A goal will be to obtain a number of candidate > genes containing repeat length variation as a response to selectional > regimes. Another goal will be to build models for how repeat length > variation within the protein affects structural properties. > > The project as a whole consists of three interconnected work packages. The > successful candidate will work on the work package entitled "Genomic > repeat architecture in Arabidopsis and Atlantic cod and length variation > in simple repeats" led by professor Kjetill S Jakobsen. The project has > international collaborators and research visits to collaborating partners > will be encouraged. > > Requirements/qualifications: > > The Faculty of Mathematics and Natural Sciences has a strategic ambition > of being a leading research faculty. Candidates for these fellowships > will be selected in accordance with this, and expected to be in the > upper segment of their class with respect to academic credentials. > > Applicants must hold a Master's degree or equivalent in biology, > molecular biology or bioinformatics/biostatistics and have experience > with bioinformatics methods for analysis of high throughput sequencing > genome data. The applicant must be qualified for the doctoral program > within these disciplines. The successful applicant should preferably > have experience in using bioinformatics and computational methods in > a comparative or evolutionary setting. Experience with programs for > analyses of genomic repeats will be particularly useful. > > Candidates without a Master's degree have until 30 June, 2016 to complete > the final exam. > > The purpose of the fellowship is research training leading to the > successful completion of a PhD degree. > > The fellowship requires admission to the PhD program at the Faculty of > Mathematics and Natural Sciences. The application to the PhD program must > be submitted to the department no later than two months after taking up > the position. For more information see: > > https://ift.tt/2WXezLX > > https://ift.tt/3jVU2Az > > A good command of English is required. > > Salary: > > Position code 1017, Pay grade: 50 - 57 (NOK 430 500 - 483 700 per year) > > The application must include: > > Application letter CV (summarizing education, positions and academic > work - scientific publications) Copies of educational certificates, > and transcript of records Documentation of English proficiency List of > publications and academic work that the applicant wishes to be considered > by the evaluation committee Names and contact details of 2-3 references > (name, relation to applicant, e-mail and telephone number) > Foreign applicants are advised to attach an explanation of their > University's grading system. Please remember that all documents should > be in English or a Scandinavian language. > > In accordance with the University of Oslo's equal opportunities policy, > we invite applications from all interested individuals regardless of > gender or ethnicity. > > UiO has an agreement for all employees, aiming to secure rights to > research results a.o. > > Region: Oslo Job type: Contract Working hours: Full-time Working days: > Day Application deadline: 15 April, 2016 Location: Oslo Reference number: > 2016/3424 Home page: https://ift.tt/3lbMglt Contacts: Professor Kjetill > S. Jakobsen Telephone: +47 22854602 Associate Professor Melinka A. Butenko > Telephone: +47 22854573 Questions regarding Easycruit, contact HR Officer > Torunn Standal Guttormsen Telephone: +47 22 85 42 72 > > > > [email protected] >
via IFTTT
0 notes
Last Seen Blogs

a-vicious-faithless-angel
Freedom At Last

prologictechno
Prologic Technologies

sevmeyelim
Kötü Biriyim

lilygeb
hello!!!

megan-sandra
Megan Sandra Durbin