#plexiform neurofibromas
Text
About Neurofibromitosis
Hi. I’m Pepper. I’m a 20-something and I have a genetic disorder called Nf1, or Neurofibromitosis type 1. What the hell is neurofibromitosis? I’ll tell you! There are 3 different types, but I will be covering Nf1 only.
So, cells naturally like to divide and replicate, right? That’s what they do. 6th grade biology 101. But obviously, cells dividing and replicating everywhere unmitigated would be a bad thing, because that’s how you get tumors, cancers, etc.
So. Your body has a gene, called the Nf1 gene, that produces a chemical called neurofibromin. Neurofibromin is a tumor-inhibitor. So your body basically has a natural gene that says “no tumors”.
My body does’t have that gene! So my body says “yes tumors”!!! Which means that my body spontaneously grows thousands of tiny tumors wherever and whenever it wants. For this reason I joke that Nf1 is Yes Tumors Disease.
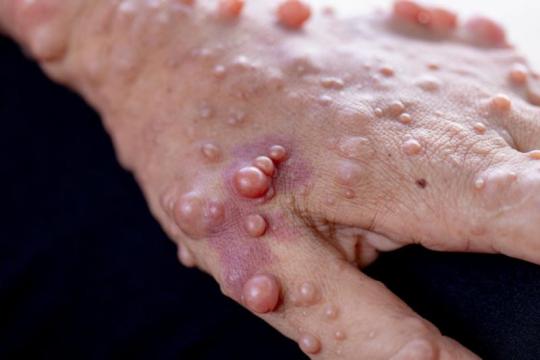
Some of these tumors are about the size of a pea or a pinhead and some are only a little bigger than this period . They might resemble a skin tag or a pimple. Those are called cutaneous neurofibromas, and they grow anywhere on the surface of the skin. They do not go away once they’re there, and there aren’t a lot of options to have them removed. But some of them are more of what you might think of as a traditional tumor - fleshy, large masses that grow and become deeply enmeshed in the nerves, and can become cancerous. These are called plexiform tumors and they can grow anywhere, including the face. I have three of them; I had surgeries to remove two of them that were not successful, and the third isn’t at the size or rate of growth where I’d be considering surgery yet, especially given that it’s on my neck and it would be a complicated surgery. Brain tumors are common with Nf1 too; I myself have a stable brain tumor, an optic glioma, so I need to get annual or biannual MRIs for... the rest of my life, essentially, to make sure that tumor doesn’t grow and new ones don’t appear.
Tumor growth is related to hormones, and one may go through periods of their life where no tumors grow, followed by periods of their life where tumors grow extremely frequently. I didn’t get a single neurofibroma until I was 22, but now that I’m in my mid-20s I have to check my body nearly daily to keep track of when and where new ones are showing up.
Nf1 affects every organ and system in the body and comes with a host of other symptoms, including bone deformities, learning disabilities, and problems with vision. It is the most common genetic disorder in humans, affecting about 1 in 2500 people. It’s autosomnal dominant, meaning a patient with Nf1 has a 50% chance of passing the condition onto their child, if their partner does not have Nf1. If their partner also has Nf1, it’s a 100% chance. For this reason, I have made the personal decision not to carry a child.
One of the main ways that Nf1 affects me (other than the tumors, duh, which absolutely suck) is scoliosis. I developed severe scoliosis at the age of 6 and needed to have back surgery at Johns Hopkins by the age of 8, to fuse my spine with metal rods and screws so it wouldn’t continue to curve. I then had surgery to fuse additional vertebrae at age 12, and a surgery at 22 to replace hardware that had cracked. I’m fused T2-T12, all but one of my thoracic vertabrae, so half of my spine does not bend. I physically cannot slouch. I set off metal detectors everywhere and I always get patted down by TSA. I have ramrod straight posture with no effort put in, which is sometimes a plus! Spinal fusions are common in Nf1 patients and the more vertebrae are fused, the less mobility you have. I am lucky that I am still able to twist and bend, though I can’t touch my toes and I’m supposed to squat instead of bending over.
Here’s a Johns Hopkins page where you can learn more :) Though please ask me any questions if you’re curious! I love getting to talk to people about my disorder and I don’t find questions offensive. Nf1 is a fascinating condition. And if you have nf1, please reach out to me! I’ve never had a friend with my disorder and it’s an unimaginably lonely feeling
32 notes
·
View notes
Text
Short Description on Plexiform Neurofibroma
Short Description on Plexiform Neurofibroma
Plexiform neurofibroma is a rare type of tumor that affects the peripheral nerves. It is a benign tumor that can occur anywhere in the body, but it is most commonly found in the head, neck, and extremities. Plexiform neurofibroma is often associated with neurofibromatosis type 1 (NF1), a genetic disorder that affects the development and growth of nerve…
View On WordPress
0 notes
Text
⚠️MUTUAL AID REQUEST⚠️
(Do not tag!!)
Hello friends, my good friend Hyde is in desperate need of funds for a wheelchair due to being severely disabled and having been bedbound for 8 years. He has Neurofibromatosis type-1, which has also resulted in severe scoliosis that extensive surgery has been unable to fix yet. His mother also has rheumatoid arthritis, and is the sole caregiver for him and his sister, who is intellectually disabled from S4 brain cancer.
It would mean a lot to him if you could share this around and/or donate, he needs all the help he can get.
You can donate to either of these links:
Kofi @ Vampire
Give a Little @ Hydeyfund
Learn more about NF-1 here:
NCBI.gov - Plexiform Neurofibroma
Cancer.net - Neurofibromatosis Type-1
Goal 1: $9,000 (Basic wheelchair)
Goal 2: $18,000 (Power assist and modifications)
Goal 3: $24,000+ (Motorized and all terrain wheels)
Currently: 195/18,000
141 notes
·
View notes
Photo

“Why do we need Disability Pride?”
A comic I made for Disability Pride. Under the keep reading is an in-depth image description of the comic.
[description one: a seven-panel comic titled ‘Why do we need Disability Pride?’ where two ignorant able-bodied people have their misconceptions about disability challenged. at the end of the comic, they are involved in the community.
[description two: A text at the beginning of the comic reads, “Why do we need Disability Pride?” Beneath that is a balanced scale with two people sitting on either side. On one side is Ekam, a Sikh man wearing a turban, button-up, and khakis. Above it reads, “Ekam is uncomfortable around disabled people.” On the other side of the scale is Bailey, a fat white woman wearing a long-sleeved shirt, tights, and a carrier purse which are all purple. Above her it reads, “Bailey thinks every disabled person is an inspiration.” To either side of them are panels providing examples of how their ableism manifests.
To the left of Ekam is a panel of him speaking to a coworker, Alazaye, a fat Black woman with half-up hair buns in a wheelchair wearing a tank top and jean shorts. Ekam looks unsettled, but polite. In a speech bubble above him he says, “So.. uh,... hey..” Imposed over-top of his dialogue is Alazaye’s thought bubble that reads, “he’s only awkward around me..” while looking away, frustrated.
To the right of Bailey is a panel of her speaking to a stranger, Caden, a white man with facial and limb differences due to burns using arm crutches. He is wearing a t-shirt and long-sleeved shirt with jeans. Bailey is touching his shoulder comfortingly while saying, “it’s so BRAVE of you to be OUTSIDE!” Caden is looking at her directly while scowling, but not speaking.
In the next panel is a landscape of a Disability Pride parade. Two people in mobility scooters are leading the parade while carrying a banner of zigzag Disability Pride Flag. On the left side of the banner is a fat East Asian woman wearing a blouse with her hair up. On the right is a dark-skinned Black woman with locs, self-harm scars, and Plexiform neurofibromas on her face wearing a bucket hat and t-shirt. Around them are other disabled people and allies carrying signs or celebrating in the street. Above the panel it reads, “Disability Prides in America have been held since 1990 when the ADA was signed.” Below it reads, “The work of disabled activists during the month encourages others to break down bias they may have towards those with disabilities.”
In the next panel Ekam approaches a group of people within the parade, smiling and waving nervously. In the foreground of the illustration is Ruben, a South Asian enby waving to Ekam with one of their prosthetic arms. Caden from before is standing beside Ekam wearing a sunhat to protect his eyes. Below the panel it reads, “Be brave and reach out.”
In the next panel Bailey listens to a white redheaded woman wearing goth clothes in a wheelchair speak while Ernie, a chubby Black man with a high-top fade wearing an autistic infinity t-shirt and jeans, is prompting Bailey to listen to the other. Above the panel it reads, “Really listen to what we have to say.”
In the final panel, Ekam and Bailey are engaging with and listening to their disabled friends at the parade. Ekam is telling a joke to Ruben in the left corner of the panel, Caden is listening to Ernie infodump about his special interest in the middle, and to the right Alazaye is joined hands with Bailey while looking at each other romantically. Above the group it reads, “You’ll begin to see what we’ve been trying to show you.” Below the group it reads, “WE ARE DISABLED!” Then outside the panel it reads in italics, “NOT DISPOSABLE.”
Thank you for reading. Happy Disability Pride!
#disability#disability pride#image transcribed#disabled rights#mobility devices#disability pride month#face difference#cw self harm scars#burn survivor#mobility scooter#autism#disabled not disposable#wheelchair user#tw self harm scars#plexiform neurofibromas#physically disabled#accessibility#image described#comic#my art#daly art
4K notes
·
View notes

Text
Lupine Publishers | Micro-Environmental Systems and Endothelial Cells in Cooperative Tumorigenesis Account for Potential Malignant Transformation in Neurofibromatosis Type 1 Patients
Open Access Journal of Oncology and Medicine (OAJOM)

Lupine Publishers Open Access Journal of Oncology and Medicine (OAJOM)
Abstract
Overall tumorigenesis in neurofibromatosis type 1 patients constitutes a series of specific targeting events with a central role enacted by proliferation of fibroblasts and endothelial cells in overproduction of growth factors and cytokines such as transforming growth factor-beta and CXCL12 cytokine. The plexiform neurofibroma well-illustrates dimensions of such cooperative participation within operative fields of the initial Schwann cell proliferation leading in a significant number of patients to malignant transformation of the peripheral nerve sheath tumors. Inclusive directions in operative targeting of Schwann cells or astrocytes are staged performance in the transformation of hyperproliferative induction and constitute further evolutionarily defined incorporation of such systems as endothelial cells. Hyperproliferative cell subsets are initial and also consequential target formulation of potential malignant states as induced in malignant peripheral nerve sheath tumors.
Introduction
Neurofibromatosis type 1 (NF1) is a neurogenetic disorder and involves both heterozygous and homozygous absence/reduction of neurofibromin that acts normally as a tumor suppressor. There is a need to assess predisposing genetic factors and loss of heterozygosity causing emergence of aggressive neoplasms in patients with NF1 [1]. The two hit hypothesis helps account for the emergence of Schwann cell-based proliferations and for neurofibromas and plexiform neurofibromas. Gherkin may act on tumorigenesis of cutaneous neurofibromas via growth hormone secretagogue receptor [2]. It is important to consider the neurofibroma that is based on micro-environmental potentiation of tumor generation in patients that develop malignant nerve sheath tumors and astrocytomas in patients with NF1 +/- genotype; this occurs in a manner that involves growth factor overactivity and mast cell and endothelial overactivity within a milieu that dysfunctionally stimulates tumorigenesis. Reactive oxygen species overproduction lead to epithelial-mesenchymal transit in patients with neurofibromin deficiency and plays a crucial role in NF1 tumor growth [3]. RAS activation alone is not sufficient for malignant transformation of peripheral nerve sheath tumors; signal transduction may potentially help identify therapies for this neoplasm type [4].
Neurofibromin
The dynamics of neurofibromin as a cytoplasmic protein involve the regulation of K-Ras, and the PI3K/Akt pathways; absence of neurofibromin leads to overactivation of these pathways in various ways in inducing tumorigenesis in such lesions as optic tract pilocytic astrocytomas, brain stem astrocytomas and also other CNS astrocytomas in terms of progression of these lesions. The cell of origin determines the temporal course of neurofibromatosis-1 low-grade glioma formation [5]. The micro-environment of plexiform neurofibromas of peripheral nerves and of nerve plexi include a 10% risk of malignant change with subsequent aggressive clinical behavior in the affected patients. Over expression of cellular retinoid acid binding protein 2 is reported in several cancer types, including malignant peripheral nerve sheath tumors (MPNSTs) [6].
Related Tumor Predispositions
The neurofibromin insufficiency status in Schwann cells and fibroblasts allows for enhanced participation of immune system component cells such as microglia as evidenced in optic pathway low-grade astrocytomas. Telomere erosion is described in many tumor types and may potentially drive genomic instability and clonal progression in NF1-associated MPNSTs [7]. Tumor dimensions include proliferation of astrocytic cells in optic pathways, and of various subtypes of stromal cells such as fibroblasts and mast cells in the peripheral nervous system. It is significant to consider particularly the micro-environmental active participation in the genesis of the most common tumor type in Neurofibromatosis type 1 patient, that is the neurofibroma, which invokes proliferation of fibroblasts and endothelial cells. The congenital plexiform neurofibroma is in fact a hypervascular lesion that transgresses tissue margins and induces a significant risk for malignant transformation. NF1 loss is the primary driver of tumorigenesis in neurofibromatosis type 1-related plexiform neurofibroma [8]. It is further to such considerations that important cooperative intervention in malignant transformation of plexiform neurofibromas invokes multi-type cells in inducing proliferation of an integral Schwann cell-fibroblastic twin population in enhancing potential malignant transformation of the peripheral nerve sheath. A therapeutic window for neuroprotective intervention exists as detected by optical coherence tomography in mice with optic glioma, and particularly as an accurate biomarker of retinal ganglion cell apoptosis [9]. The heterozygous absence of one neurofibromin allele in mice results in plexiform neurofibromas and low-grade optic pathway astrocytomas. Mast cells appear to play a causal role in neurofibroma formation and also in microglia in optic pathway glioma evolution [10]. Such implications of the micro-enviromental factors includes a distinctive cooperative participation that carries implications for significant enhancement of cell proliferation and of such cytokines such as transforming growth factor and CXCL12 in formulating malignant transformation in such tumors. The methylemetetrahydrofolate reductase 1298 and 677 gene polymorphisms are related to optic glioma and hamartoma risk in NF1 patients through effects on DNA synthesis and methylation [11].
Convergent Targeting
The related tuberous sclerosis complex is analogous to neurofibromatosis type 1 as a neurogenetic disorder associated with increased risk for astrocytomas in the form of subependymal giant cell astrocytomas. A convergent targeting of systems of cell proliferation include in particular cyclic AMP and Ras in a manner that includes dimensions of micro-environmental conditioning. Mutations of the NF 1 gene are frequent in many cancer types in patients without NF1 and this is suggestive of a more general role for the NF1 gene in oncogenesis. In melanoma NF1 mutations potentially drive tumorigensis and promote drug resistance [12]. Inclusive dynamics allow for permissive tumorigenesis in a manner that includes the incorporation of malignant transformation within confines of a Schwann cell-fibroblast-endothelial cell system in the case of malignant peripheral nerve sheath tumors. Astrocytes and microglia are analogous counterparts in the induction of CNS astrocytomas. Such considerations are inclusive phenomena of multi-component induction of potential malignancy that recharacterizes conditioning of the micro-environment of proliferative states preceding tumorigenesis. Interaction between neoplastic Schwann cells and their surrounding neural microenvironment has important implications for early cellular events promoting tumorigenesis in neurofibroma development [13].
Performance Dynamics
Performance dynamics of tumors in neurofibromatosis type 1 may potentially modify the biologic significance of a two-hit hypothesis in a manner that implicates micro-environmental conditioning of the resultant cell hyperplasias and proliferations in such lesions as peripheral nerve sheath tumors and astrocytomas. NF1 provides unique vantage points to examine co-contributions of molecular, cellular, and tissue processes in tumor biology [14]. Such proposed dimensions invoke in particular an over-activation in production and action of growth factors that provoke selective malignant transformation of hyper-proliferative lesions composed of Schwann cells and astrocytes in the peripheral and central nervous systems respectively. Plasma soluble levels of transforming growth factor-beta and interleukin-6 are increased in NF1 patients and a shift towards an anti0inflammatory profile has been reported in cells expressing cytokines [15].
Hyperproliferation
The hyperproliferative states affecting Schwann cells and astrocytes invoke also fibroblast and microglial cell proliferations in a manner transforming tumorigenesis. Such facilitation to tumorigenesis invokes dimensions of transformation as well seen in plexiform neurofibromas that may undergo malignant transformation in a significant number of affected individuals. Such considerations are selective targeting of specific cell subpopulations in a manner that allows permissive transformation. Insertional mutagenesis identifies a STAT3/Arid1b/beta-catenin pathway that drives neurofibroma initiation in the context of Nf1 loss [16]. Mast cells and fibroblasts may potentially incorporate endothelial cells that may participate as central dysregulatory dimensions in plexiform neurofibroma tumorigenesis. The provocations for malignant transformation further cooperate in systems of derivative consequence as hypervascular lesions that subsequently lead to potential malignant cells in individual patients. Cross species comparative oncogenomic may identify driver mutations in mouse cancer models and allow validation in human tumors [17].
Concluding Remarks
Propositional implications in tumorigenesis include the multi-component participation of Schwann cells on the one hand and of fibroblasts, mast cells, endothelial cells and also of microglia in an inductive process that includes specific pathways of malignant transformation. Endothelial cell proliferation is related to substantial participation in modes related to key-events of increased proliferation of Schwann cells and astrocytes in initial stages of lesion infliction. Inclusive phenomena have thus become systems of consequence in affecting such specific cell proliferative states. Such events occur within the added dimensions of directed targeting of multiple-agent micro environmental modeling of the initial proliferation of the Schwann cells or astrocytes. A pivotal series of roles played by fibroblasts, endothelial cells, mast cells and of microglia and astrocytes appears a dynamic milieu within added consequences of malignant transformation of both Schwann cells and astrocytes that progress as cooperative systems of tumorigenesis.
For more Lupine Publishers Open Access Journals Please visit our website: h
http://lupinepublishers.us/
For more Journal of Oncology and Medicine Impact Factor articles Please Click Here:
https://lupinepublishers.com/cancer-journal/index.php
To Know More About Open Access Publishers Please Click on Lupine Publishers
Follow on Linkedin : https://www.linkedin.com/company/lupinepublishers
Follow on Twitter : https://twitter.com/lupine_online
6 notes
·
View notes
Video
✅Follow ➡️ @my_name_is_dentist Plexiform Neurofibroma : Complex Reconstructive Craniofacial Surgery being planned next month. History : 2 prior surgeries www.facesurgeon.in www.drsunilrichardson.com +91943182860 _______________________________ FOLLOW : 📷 Instagram/dentistryzone 👤 Facebook/dentistry.zone 💎 Twitter/dentistryzone1 👻 Dentistryzone ▶️ YouTube/DentistryZone ⚫️ Tumblr/dentistryzone1 _______________________________ Tag your friends 👇 #dentistryzone #neurofibromatosis #neurofibromatose #neuroma #face #craniofacial #facialnerve #omfs #plasticsurgery #plasticsurgeon #surgery #surgeon #cirugia #medical #medic #medico #medicalstudent #medicalstudents #medstudent #medstundets #dental #dentalstudents #reconstructivesurgery #dentalstudent #medicine #drsunilrichardson #muscat #oman #kerala #tamilandu https://www.instagram.com/p/Btb_KT1IftI/?utm_source=ig_tumblr_share&igshid=1ehsnsv8haalu
#dentistryzone#neurofibromatosis#neurofibromatose#neuroma#face#craniofacial#facialnerve#omfs#plasticsurgery#plasticsurgeon#surgery#surgeon#cirugia#medical#medic#medico#medicalstudent#medicalstudents#medstudent#medstundets#dental#dentalstudents#reconstructivesurgery#dentalstudent#medicine#drsunilrichardson#muscat#oman#kerala#tamilandu
2 notes
·
View notes
Text
Neurofibromatosis Treatment Drugs Market : Industry Trends and Forecast
Neurofibromatosis type 1is a devastating innate condition that can cause clinical issues like twisting, motor brokenness, torture, aeronautics course brokenness, visual impedance and bladder or entrail brokenness. Neurofibromatosis type 2 is connected with vestibular schwannomas, or noncancerous tumors along the nerves in the frontal cortex that are locked in with hearing and harmony. Lately, in July 2021, an investigation in the U.S. exhibited that the circulatory strain drug losartan may assist patients with neurofibromatosis type 2. The assessment was aimed at Massachusetts General Hospital and Massachusetts Eye and Ear. Basically in October 2019, SpringWorks Therapeutics, Inc., a clinical-stage biopharmaceutical association, announced that the fundamental patient has been dosed in the Phase 2b ReNeu clinical starter surveying mirdametinib, an oral, little molecule expected to thwart MEK1 and MEK2, in kids and adult patients with neurofibromatosis type 1-related plexiform neurofibromas (NF1-PN). Thus, R&D in neurofibromatosis treatment is depended upon to drive advancement of the neurofibromatosis treatment drugs market.
The neurofibromatosis treatment drugs market in Asia Pacific is driven by underwriting and dispatch of new drugs. For instance, in July 2020, AstraZeneca was permitted transient prescription task for selumetinib in Japan for the treatment of neurofibromatosis type 1. Moreover, government premium in R&D of neurofibromatosis is similarly expected to help improvement of the market in Asia Pacific. For instance, in February 2021, The Australian Government proclaimed to contribute US$ 8 million to help the Children's Tumor Foundation and assessment into neurofibromatosis.
Read More : https://bit.ly/3CSq0F8
0 notes
Text
Brachial Plexus Neurofibroma Treated with Volumetrically Modulated Arc Therapy (VMAT): A Case Report

Abstract
Neurofibromatosis was first described in 1882 by Friedrich Daniel von Reckling hausen, a German pathologist. Neurofibroma is a benign peripheral nerve sheath tumor that consists of Schwann cells, associated or unassociated with axons, perineural cells, and fibroblasts. Whenever possible, the treatment of choice should be surgical, but the management depends on the location and growth pattern. We present the case of a patient with left axillary neurofibroma without neurofibromatosis (NF) in whom surgery was delayed due to involvement of the brachial plexus, so was sent to radiotherapy, planned and treated with volumetrically modulated arc therapy (VMAT).
Keywords: Neurofibroma; VMAT; Radiotherapy; Axillary Tumor; INCART
Abbreviations: NF: Neurofibromatosis; VMAT: Volumetrically Modulated Arc Therapy; CRO: Radiation Oncology Center; INCART: Institute Rosa Emilia Sánchez Pérez de Tavares; CW: Clockwise; NTO: Normal Tissue Objective; OAR: Organ at Risk
Introduction
Neurofibromas are benign tumors that arise from nerves and are composed of Schwann cells, perineural cells, and fibroblasts [1]; typically arise in the setting of neurofibromatosis type I (NF I) [2], but may also occur independently, when it comes to multiple tumors is the main clinical manifestation of the different types of neurofibromatosis and are processes that are transmitted with autosomal dominant inheritance with variable penetrance. In most cases are treated by surgery, especially when they are symptomatic or with evident anatomical changes [3,4], but when a neurofibroma involves a particularly long segment of nerve or nerves, are generally impossible to remove without removing the entire nerve, causing a major neurological deficit, therefore, these variants are not usually subjected to surgery. In this report, we describe an axillar neurofibroma in a patient without NF I, which was treated with radiotherapy using volumetrically modulated arc therapy (VMAT).
Case Summary
Female patient, 55 years old, who had previously presented to an external surgery department was remitted to the Radiation Oncology Center (CRO) at National Cancer Institute Rosa Emilia Sánchez Pérez de Tavares (INCART), in Santo Domingo, Dominican Republic, with history of pain and volume increase in left armpit, diagnosed as non-surgical axillar neurofibroma. Medical record, including MRI, CT and pathologic specimen, were reviewed. The patient was considered initially for surgery like standard treatment, but during process evidenced brachial plexus commitment, because of this was omitted the surgery and was referred to radiation oncology. The Physical exam reported: Karnofsky (KPS) 100%, ECOG 0, in the left axillary region was felt a mass about 10 cm in diameter (Figure 1), mobile, painful, hard consistency with left arm weakness. A shoulder MRI with gadolinium reported: oval image 8.5 x 5.3 cm anteroposterior, anterior to the subclavian neurovascular bundle located in the anatomical path of the brachial plexus, highly suggestive of neural image type neurofibroma. Described image shows a bottom center 1.9 cm cystic degeneration, in turn shows an intense post gadolinium enhancement and heterogeneous accordance with important vascularization (Figure 2).
Simulation Ct and delineation
The patient was positioned in supine using a table for breast carbon fiber of the CIVCO® brand. Package tumor localization (TumorLOC®) Brilliance CT was used for the location of tumor tomography simulation. This tool allows you to place the isocenter manually depending on the selected organs and can create orthogonal beams associated with that default position of isocenter. Tomographic scanning technique was 120kV, axial cut, over 200,3 mm apart.
Field arrangement and treatment plan
The treatment technique corresponded to volumetrically modulated arc therapy (VMAT) for this, two semi-arches were used. The first from 0° to 179° towards the direction of clockwise (CW) and the second reverse direction arc (179° to 0°). They were established for each arc 98 control points with maximum speed of gantry 4.8 deg / s. Each semi-arch lasted 48 seconds (0.8min) (Figure 3 & 4). The bodies’ risks were considered for planning spinal cord, left lung, left humeral head and left mammary gland. As establishment of an NTO (normal tissue objective) use the automatic option. Resolutions (mm) and priorities (%) planning for OAR (organ at risk) were, respectively: spinal cord 2 mm and 35%, humeral head 2.65 mm and 25%, left lung 3 mm and 30%, left breast 3 mm and 25%.
Assessment treatment plan
Evaluating different plane cuts ensure that 100% of the prescribe dose is within the volume of total dose planning 5940 cGy. The bodies assessed risks are below the limits set by Quantec / RTOG (Figure 5).
One of the benefits of this proposal lies among other things in the runtime patient treatment compared to some other CRT3D or IMRT technique. The maxim and average doses to OAR were: 27.3 Gy / 10.5 Gy (VMAT) and 36 Gy / 9.12 Gy (IMRT)for left humeral head (green); 60.0 Gy / 5.2 Gy (VMAT) and 60.5 Gy / 5.7 Gy (IMRT) for left breast (yellow); 8.5 Gy / 2.7 Gy (VMAT) and 9.6 Gy / 2.9 Gy (IMRT)for spinal cord(purple) and finally 17.3 Gy / 1.77 Gy (VMAT) and 35.5 Gy / 2.5 Gy (IMRT) for left lung (blue) (Figure 6)
QA
The two semi-arches were evaluated for creep refers using an amorphous silicon system to perform dosimetry portal. Descriptors parameters for evaluation were the index gamma 3% and distance agreement (DTA) 3mm. Predicted doses vs. the portals doses obtained successfully passed the evaluation criteria (Figure 7).
Diagnosis
Left axillary neurofibroma. The microscopic biopsy description reported: sections showed mesenchymal neoplasm, partially encapsulated, constituted by the proliferation of spindle cells, elongated nuclei arranged in swirling pattern and fibrillar collagenous stroma storiform on congestive vessels with thickened walls and areas of bleeding. Differential diagnoses included: schwannoma, malignant peripheral nerve sheath tumor and desmoid tumor [5].
Discussion
Neurofibromatosis was first described in 1882 by Friedrich Daniel von Recklinghausen, a German pathologist. It is a disease caused by an abnormality in a gene on chromosome 17.Thediagnosis of Neurofibromatosis type I (NF1) is confirmed If 2 or more of these criteria are met in an individual: six or more stains coffee with milk greater than 5 mm in pre pubertal patients and over 15 mm in post pubertal patients; two or more neurofibromas of any type or one plexiform neurofibroma; sign Crowe (axillary or inguinal freckling); [6-9] Glioma optic nerve; two or more Lisch nodules (iris harmartomas); typical bone lesions (dysplasia of the sphenoid dysplasia or thinning of long bone cortex with or without pseudoarthrosis); history of neurofibromatosis type I in parents or siblings [10]. Meanwhile, to make the diagnosis of NF 2 is necessary to have the following criteria: bilateral masses in the eighth cranial demonstrated techniques by appropriate images or first-degree relative with NF2 and one of the following: unilateral mass the eighth cranial nerve, or two of the following: neurofibroma, meningioma, glioma, schwannoma, subcapsular lenticular opacity youth back [10].
In the case of our patient, it has none of the criteria for NF1or 2. Neurofibroma is a benign peripheral nerve sheath tumor that consists of Schwann cells, associated or unassociated with axons, perineural cells, and fibroblasts. The tumor routinely expresses S-100 protein. Benign neurofibromas most often occur in association with neurofibromatosis type 1 (NF1) but may also occur sporadically [1,2], like in our case report.
The biological behavior of neurofibromas is usually benign, but it is not rare sarcomatous degeneration in plexiform. Neurofibromas occur in multiple locations, exhibit different growth patterns and morphologies at highly variable rates, a classification system stratifies these tumors into 5 types: localized cutaneous, diffuse cutaneous, localized intraneural, plexiform, and massive soft tissue neurofibromas [4].
The management depends on the location and growth pattern. Whenever possible, the treatment of choice should be surgical, especially when they produce functional or cosmetic déficits, ideally with complete removal of the tumor, but some tumors diffusely involve adjacent normal tissues such as nerves and blood vessels, making complete remove of the tumoral most impossible [1,3,4,6,8,9]. Medical therapies have been investigated, most are still investigational [4] and Radiotherapy has been rarely used like initial treatment, but by default in patients with incomplete resection [1,4]. Needle et al. [6] reported 121 children with plexiform neurofibromas who underwent resection and were followed for over 20 years. Those that were entirely removed were less likely to recur and had a longer median time to failure. Near-total and subtotal resections demonstrated recurrence rates of 39.5% and 44%, respectively.
Conclusion
We must emphasize team work and multi disciplinary approach to tackling the disease. The commitment caused by neurofibromas depends in much of their location, skin lesions usually cause deformity, while more injuries shallow tend to generate functional compromise. Despite being mostly benign, some cause destruction secondary to the pressure exerted, so the symptoms are depend on the size of the lesion. The therapeutic approach should be multidisciplinary, with the initial treatment recommended surgery, but in case of not being able to perform or partial resection, radiotherapy with doses between 54-60 Gy can achieve adequate local control. In reviewing the literature an extensive description of the disease and epidemiological data was found are scarce, so studies to know the disease more accurately are needed.
To Know More About Cancer Therapy & Oncology International Journal Please click on:
https://juniperpublishers.com/ctoij/index.php
For more Open Access Journals in Juniper Publishers please click on:
https://juniperpublishers.com/index.php
For more about Juniper Publishers Please click on:
https://juniperpublishers0.wixsite.com/juniperpublishers
#Juniper publishers#Open access Journals#Peer review journal#Juniper publisher reviews#Juniper publisher journals
0 notes
Text
Selumetinib
In this article, we will discuss Selumetinib (Dosage Overview). So, let’s get started.
IndicationsSelumetinib is indicated for the treatment of pediatric patients 2 years of age and older with neurofibromatosis type 1 (NF1) who have symptomatic, inoperable plexiform neurofibromas (PN).DosageRecommended DosageThe recommended dosage of Selumetinib is 25 mg/m² orally twice daily (approximately…
View On WordPress
0 notes
Text
I've been sad about my body recently. I have a large Plexiform Neurofibroma on my chest and it's heavily disfigured me. I try and ignore it but idk it's just making me self conscious latley. I bind my chest anyway when I go out so that kinda hides it. However I still see it and feel it other times. It makes me feel ugly. Living with Nf1 is tough.
1 note
·
View note
Text
Best Neurologist in Pitampura - What is Neurofibromatosis Type 1
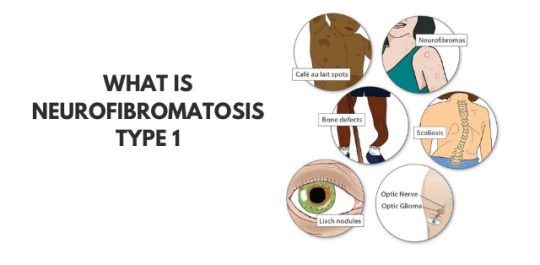
Dr Shailesh Jain - Neurofibromatosis type 1 is characterized by changes in skin color and also the development of tumors along the veins within the skin, brain, and other parts of the body. The signs and symptoms of this condition are widely different among affected people.
Neurofibromatosis type 1 also called as Recklinghausen disease or von Recklinghausen disease or von Recklinghausen’s phakomatosis or von Recklinghausen’s neurofibromatosis or neurofibroma (multiple) or peripheral neurofibromatosis.
At birth or early childhood, affected individuals may have relatively large, benign tumors that consist of bundles of nerves and other tissue (plexiform neurofibromas). Individuals with Neurofibromatosis type 1 may also develop benign nodules on the colored regions of the eyes (Lisch nodules), or tumors in the nerves of the visual pathway (optic pathway gliomas) by Best doctor for brain stroke in Shalimar Bagh.
According to Dr Shailesh Jain, Neurofibromatosis type 1 could also be characterized by unusually large head size (macrocephaly) and comparatively small height. Additional abnormalities may be present, like episodes of uncontrolled electrical activity within the brain (seizures), Learning disabilities, and lack of attention; Speech difficulties; Abnormally increased activity (hyperactivity), And skeletal malfunctions, including progressive curvature of the spine (scoliosis), bending of the lower legs (pseudoarthrosis) and improper development of some bones.
Symptoms of Neurofibromatosis type 1
Flat, brown spots (cafe au lait) on the skin -These harmless spots are common in many folks. Having more than six cafe au lait spots suggests Neurofibromatosis type 1. they're usually present at birth or appear during the primary years of life. After childhood, new spots stop appearing.
Freckling in the armpits or groin area - Freckling usually appears by ages 3 to 5 Freckles are smaller than cafe-au-lait spots and tend to occur in clusters in skin folds.
Tiny bumps on the eye's iris (lynch nodules) - These harmless nodules aren’t easily seen and don't affect vision.
Bone deformity - Abnormal bone development and reduction in bone mineral density can lead to bone deformities such as curved spine (scoliosis) or tilted lower leg.
Tumor on the optic nerve (optic glioma) - These tumors usually appear at the age of 3, rarely in childhood and adolescence, and almost never in adults.
Larger than average head size - Children with Neurofibromatosis type 1 are larger than the average head size due to increased brain volume.
short stature - Children who have Neurofibromatosis type 1 often have lower-than-average height.
Causes of Neurofibromatosis type 1
The Neurofibromatosis type 1(NF1) gene on chromosome 17 produces a protein called neurofibromin that regulates the growth of your cells. Mutation of this gene causes neurofibromin loss and uncontrolled cell growth.
NF1 complications
Neurological problems - Thinking and learning difficulties are the most common neurological problems associated with NF1. Unusual complications include epilepsy and the creation of excess fluid in the brain.
Vision problems - Sometimes a tumor develops on the optic nerve, which can affect vision.
Heart problems - People who have NF1 have an increased risk of hypertension and blood vessel abnormalities may develop.
Trouble breathing - Rarely, plexiform neurofibromas can exert pressure on the airway.
About Dr. Shailesh Jain, Neurosurgeon, Neurologist
Dr. Shailesh Jain is the Best neurologist in pitampura and Shalimarbagh has been performing with excellent results for the last 16 years. He has vast experience in this field. Dr. Shailesh Jain runs his Arihant Neurospin Clinic in Pitampura and Max Superspeciality Hospital, Shalimar Bagh. Dr Shailesh Jain is Principal Consultant Neurosurgery and Neurointervention at Max Hospital Shalimar Bagh and runs his own Arihant Neurospine clinic in Pitampura and Shalimar Bagh.
#Dr shailesh jain AIIMS Neurosurgeon#Best doctor for brain stroke in pitampura#Best neurologist in pitampura#Top Neurosurgeon in pitampura#Best doctor for back pain in pitampura#Best doctor for headache in pitampura#Best doctor for migraine in pitampura#Best doctor for fits in pitampura#Best doctor for brain hemorrhage in pitampura#Best doctor for depression in pitampura#Best doctor for Stroke Intervention in pitampura#best doctor for head and spine injury in pitampura#Best doctor for epilepsy in pitampura
0 notes
Text
CHMP backs Roche’s Enspryng for rare nerve disease NMOSD

Roche’s Enspryng has been recommended for approval in the EU for treating neuromyelitis optica spectrum disorder (NMOSD), extending the treatment options for people with the life-threatening rare disease.
The CHMP has backed the drug in patients aged 12 or more with NMOSD that tests positive for aquaporin-4 (AQP4) antibodies, a biomarker seen in around 80% of NMOSD cases. Enspryng was approved for the same indication in the US last year.
NMOSD is a lifelong and debilitating autoimmune disorder of the central nervous system, often misdiagnosed as multiple sclerosis, that causes damages to the optic nerves and spinal cord, leading to blindness, muscle weakness and paralysis. It affects roughly one to two people per 100,000 in the EU.
IL-6 inhibitor Enspryng isn’t the first drug to treat NMOSD to get a green light from the CHMP, as Alexion’s complement C5 blocker Soliris (eculizumab) has been approved for the disease in the EU since 2019. However, according to Roche is the only therapy that can be used in adolescents over 12 as well as adults. Alexion has phase 3 trials on the go in children and adolescents with NMOSD.
The Swiss pharma group also says its product is the only subcutaneous treatment option for NMOSD that can be administered at home every four weeks, after an initial three injections given two weeks apart. Soliris is given by intravenous infusion every two weeks, after an initial period of weekly dosing.
Another NMOSD therapy, Horizon Pharma/Viela Bio’s Uplizna (inebilizumab), has been approved in the US but isn’t yet available in Europe. It too is administered by intravenous infusion.
Meanwhile, Alexion’s follow-up to Soliris – Ultomiris (ravulizumab) – is also in late-stage clinical development as an infusion for NMOSD, although the drugmaker is also working on a subcutaneous formulation of the antibody. Also back in early-stage development is an oral therapy based on cladribine developed by Swiss biotech Chord Therapeutics.
Enspryng’s approval based on two clinical trials which showed that the rate of relapse with Enspryng was a quarter of that in a placebo group, both given on top of other immunosuppressive drugs.
Roche’s drug was one of several new medicines recommended for approval at the CHMP’s April meeting.
Regeneron’s first-in-class ANGPTL3 inhibitor Evkeeza (evinacumab) got a positive opinion for homozygous familial hypercholesterolemia (HoFH), a genetic form of high cholesterol, in patients aged 12 and over, and the committee also backed AstraZeneca’s MEK 1/2 inhibitor Koselugo (selumetinib) for children with neurofibromatosis type 1 (NF1) plexiform neurofibromas (PN).
Leo Pharma’s IL-13 inhibitor Adtralza (tralokinumab) got the go-ahead as a treatment for adults with moderate to severe atopic dermatitis, as did Celgene’s Onureg (azacitidine) as a maintenance therapy for patients with acute myeloid leukaemia (AML).
Other highlights included an extension of the Indications for AbbVie’s Venclyxto (venetoclax) as a first-line treatment – in combination with azacytidine or decitabine – in newly-diagnosed AML patients ineligible for intensive chemotherapy.
AZ also got the go-ahead for Tagrisso (osimertinib) for the adjuvant treatment after surgery of patients with early-stage EGFR-positive non-small cell lung cancer (NSCLC).
The post CHMP backs Roche’s Enspryng for rare nerve disease NMOSD appeared first on .
from https://pharmaphorum.com/news/chmp-backs-roches-enspryng-for-rare-nerve-disease-nmosd/
0 notes
Text
NEUROFIBROMATOSIS TREATMENT DRUGS MARKET ANALYSIS
Neurofibromatosis Treatment Drugs Market, by Disease Type (Neurofibromatosis 1 (NF1) and Neurofibromatosis 2 (NF2), and Schwannomatosis), by Distribution Channel (Hospital Pharmacies, Retail Pharmacies, and Online Pharmacies), and by Region (North America, Latin America, Europe, Asia Pacific, Middle East, and Africa) - Size, Share, Outlook, and Opportunity Analysis, 2020 - 2027
Press Release : Neurofibromatosis Treatment Drugs Market
Request Sample Download Pdf
There are three types of Neurofibromatosis; Neurofibromatosis type 1, Neurofibromatosis type 2, and Schwannomatosis. Neurofibromatosis type (NF) 1 is a common monogenic disorder of neurocutaneous tissue growth that arises secondary to mutations in the tumor suppressor gene NF1. It features an autosomal dominant pattern of inheritance. Individuals with NF1 typically have an increased predisposition to a variety of benign and malignant tumors and develop neurofibromas, and axillary and inguinal freckling. Neurofibromatosis 2 (NF2) is less common than NF1. Common signs and symptoms of NF 2 include benign, slow-growing tumors affecting the cranial, spinal, and peripheral nerves, as well as the meninges. KOSELUGO- selumetinib is the only drug available for the treatment of Neurofibromatosis Type-1. Moreover, some drugs such as bevacizumab are also used as off-label for the treatment of Neurofibromatosis
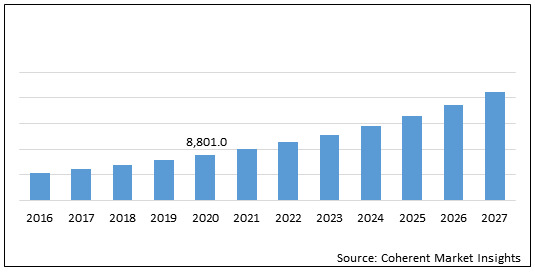
The global neurofibromatosis treatment drugs market is estimated to be valued at US$ 8,801.0 million in 2020 and is expected to exhibit a CAGR of 13.40% during the forecast period (2020-2027).
Figure 1. Global Neurofibromatosis Treatment Drugs Market Value (US$ Mn)
Global Neurofibromatosis Treatment Drugs Market: Drivers
Increasing focus of major players on research and development of novel therapies for treating neurofibromatosis. Moreover, clinical studies are well supported by regulatory authorities which is expected to drive the growth of the global neurofibromatosis treatment drugs market. For instance, in October 2019, SpringWorks Therapeutics, Inc., a clinical-stage biopharmaceutical company focused on developing life-changing medicines for patients with severe rare diseases and cancer, initiated the Phase 2b ReNeu clinical trial to evaluate the mirdametinib (formerly PD-0325901), an oral, small molecule designed to inhibit MEK1 and MEK2, in children and adult patients with neurofibromatosis type 1 (NF1)-associated plexiform neurofibromas (NF1-PN). In July 2019, the European Commission granted Orphan Drug Designation for SpringWorks Therapeutics’ mirdametinib (formerly PD-0325901), an oral, small molecule inhibitor of MEK1 and MEK2, for the treatment of neurofibromatosis type 1 (NF1).
Global Neurofibromatosis Treatment Drugs Market – Impact of Coronavirus (COVID-19) Pandemic
The coronavirus (COVID 19) pandemic and lockdown in various countries across the globe have impacted the financial status of businesses in all sectors. Private healthcare is one such sector, which has been majorly impacted by the COVID-19 pandemic.
The lockdown in various countries due to the pandemic has placed an economic burden on the private healthcare sector.
Moreover, the coronavirus pandemic has hampered the development, production, and supply of healthcare products (medical devices and medicinal products) and affected growth of healthcare businesses of various companies across the globe.
The lockdowns have led to closure of industrial establishments, except manufacturing of essential commodities, and disrupted supply chains.
The COVID-19 pandemic has affected the economy in three main ways; 1) by directly affecting the production and demand; 2) by creating disruptions in distribution channels; and 3) by its financial impact on firms and financial markets
As a result, the impact of coronavirus (COVID-19) pandemic is also expected to limit growth of the global neurofibromatosis treatment drugs market during the forecast period.
Figure 2. Global Neurofibromatosis Treatment Drugs Market Value (US$ Mn), by Region, 2020
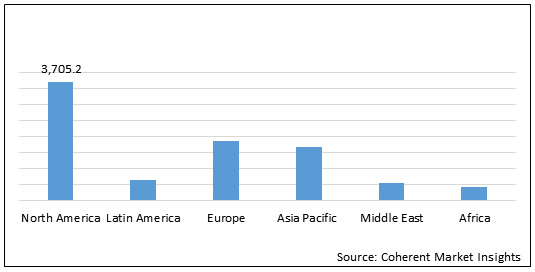
North America neurofibromatosis treatment drugs market is expected to show significant growth during the forecast period owing to presence of key players and research institutes who are involved in clinical trial programmes. For instance, on November 28, 2017, the University of Alabama at Birmingham initiated phase 2 clinical study on Binimetinib in children and adults with nf1 plexiform neurofibromas. On February 18, 2020, the University of Alabama at Birmingham initiated phase 2 clinical trial of crizotinib for children and adults with neurofibromatosis type 2 and progressive vestibular.
Key Players
Major Player operating in the global neurofibromatosis treatment drugs market is AstraZeneca Plc.
About Us:
Coherent Market Insights is a global market intelligence and consulting organization focused on assisting our plethora of clients achieve transformational growth by helping them make critical business decisions.
What we provide:
Customized Market Research Services
Industry Analysis Services
Business Consulting Services
Market Intelligence Services
Long term Engagement Model
Country Specific Analysis
Contact Us:
Mr. Shah
Coherent Market Insights Pvt. Ltd.
Address: 1001 4th ave, #3200 Seattle, WA 98154, U.S.
Phone: +1-206-701-6702
Email: [email protected]
0 notes
Text
Pleural Malignant Solitary Fibrous Tumor Mimicking a Nerve Sheath Tumor in a Patient with Neurofibromatosis Type 1

Abstract
Neurofibromatosis type 1 (NF1), formerly known as von Recklinghausen’s disease is a nervous system tumor disorder of variable expressivity. Malignant solitary fibrous tumors (SFT) are rare spindle cell soft tissue tumors. We report a case of a 35-year-old male who presented clinically with NF1 and a mass that mimicked a nerve sheath tumor radiologically, but pathologically was, in fact, a malignant solitary fibrous tumor. Based on a literature review, the association between NF1 and malignant SFT has never been published.
Keywords: Neurofibromatosis type 1; Malignant solitary fibrous tumor
Abbrevations: SFT: Solitary Fibrous Tumors; NF1: Neurofibromatosis type 1
Mini Review
Neurofibromatosis type 1 (NF1) is a multisystem disease characterized by cutaneous findings, as well as benign and malignant tumors of the nervous system. NF1 occurs in about 1 in 3,000 individuals, and about half of the affected individuals are first cases in their family [1].
A solitary fibrous tumor (SFT) is a rare spindle cell tumor that most commonly arises in the pleura. These tumors comprise only 1-2% of all soft tissue tumors, and of those, between 12 to 22% are malignant [2-3].
No link has ever been made between NF1 and a malignant SFT Conzo et al. [4-5] first described SFT in an NF1 patient, and Yamamoto, et al later reported another case, but neither case dealt with malignant SFT.
Here, we describe a patient with clinical NF1, who presented with a mass that mimicked a nerve sheath tumor, but biopsy and immunohistochemical assays showed that the correct diagnosis was of a malignant solitary fibrous tumor.
Case Presentation
In August 2015, a 35-year-old man presented from an outside hospital with chest pain, recurrent left lower lobe pneumonia and a pleural mass seen on chest radiograph. Physical exam revealed multiple café-au-lait spots on his face, back, chest, abdomen, and legs that ranged from 5mm to 15mm, as well as nodules present on the patient’s right flank, as seen in (Figure 1), and lateral to his left eyebrow. Ophthalmologic exam revealed bilateral Lisch nodules. Due to the array of clinical findings, a biopsy of the right flank nodule was performed and revealed a neurofibroma with plexiform features (Figure 2).
The chest x-ray from the outside hospital was not available at the time of writing, but, as a surrogate, scout images from the initial CT scan performed in our institution may be seen in (Figure 3), displaying the thoracic mass initially appreciated at the outside hospital. A CT scan of the thorax with contrast was performed in our institution, which showed a left posterior intrathoracic soft tissue mass with areas of enhancement, neural foraminal widening and rib splaying (Figures 4a & 5a). An MRI of the same area was performed (Figure 4b & 5b) to better show the mass in question, and a biopsy was recommended as the mass was more suggestive of a nerve sheath tumor, or, less likely, a pleural fibroma.
Ultrasound-guided fine needle aspiration and biopsy of the left pleural mass showed bland spindle cells without defined architectural pattern within a collagenous background without necrosis or mitotic activity. Immunohistochemical staining was positive for CD34, supporting a diagnosis of solitary fibrous tumor (Figure 6).
Subsequently, the pleural mass was resected and histologic evaluation showed a spindle cell lesion with biphasic morphology. The majority of the tumor showed classic solitary fibrous tumor with benign features (Figures 7a & b) with a portion of the tumor showing markedly increased cellularity, cytologic atypia and increased mitotic activity (>4/10 high power fields) diagnostic of malignant transformation (Figures 7c & d). Immunohistochemical stain for CD34 (Figure 7e) was positive, confirming the findings.
Discussion
Neurofibromatosis type 1 (NF1), also known as von Recklinghausen’s disease, is caused by an alteration of the NF1 gene, which is a tumor suppressor gene located on the long arm of chromosome 17 (17q11.2). While genetic testing is not required, it is helpful in confirming the diagnosis. The NF1 gene encodes a protein called neurofibromin, which is a member of the GTPase activating protein family. A loss of function mutation to the NF1 gene results in increased cellular proliferation that manifests as multisystemic tumorous growth. Neurofibromatosis can be diagnosed clinically if at least 2 of the 7 following criteria are fulfilled: 6 or more hyperpigmented macules, axillary or inguinal freckles, two or more neurofibromas or one plexiform neurofibroma, optic nerve glioma, two or more iris hamartomas, body dysplasia, or a first degree relative with NF1 [6].
On the other hand, SFT classically presents as a pleural mass. Klemperer and Rabin first described the tumor in the pleura in 1931 [7]. Most of the tumors are benign neoplasms that are diagnosed in adults with an age range between 50-70 years-old and a male/female ratio of approximately 1 [8-10]. Of the pleural-based SFTs, most arise from visceral pleura (80%) with the remainder arising from parietal pleura [9]. Gross examination of an SFT typically shows a smooth glistening capsule surface [9]. Histologically, most tumors are composed of collagen-forming spindle-cells, arranged in interlacing fascicles with a classic “patternless” pattern [2]. The morphologic findings are typical, though not always demonstrable in small biopsy sections; therefore the characteristic immunohistochemical positivity for CD34 is significantly helpful in separating SFT from other spindle cell neoplasms [11]. Criteria for malignancy include the presence of necrosis, increased mitotic activity, greater cellularity, and cellular polymorphism [10]. Malignant SFT is diagnosed with presence of at least one of these criteria [10]. Approximately 15-35% of SFT are malignant at the time of diagnosis with clinical characteristics of higher recurrences and lower overall survival [2]. Nonetheless, regardless of benign.
Or malignant presentation of SFT, the hallmark of treatment is large en-bloc resection of the primary tumor with close clinical surveillance for recurrences. Mediastinal lymph node dissection and adjuvant treatments such as radiotherapy and chemotherapy are not usually employed for treatment purposes, but rather as palliative options for cases of distant metastasis and locally advanced recurrence that are not amenable to surgery [2].
Historically, NF1 has been associated with various tumors, most notable of which are malignant peripheral nerve sheath tumor, rhabdomyosarcoma, and gastrointestinal stromal tumor (GIST), with an overall occurrence rate that reaches up to 5-13%, 0.5%, and 6% respectively [12-14]. However, there are no systematic reviews that link NF1 with development of SFT. Only two case reports have been published with anecdotal evidences where SFT were found concurrently in NF1 patients. Of these two reports, both patients had pathologic diagnoses of benign SFTs. Specifically, Conzo et all described a patient who had an encapsulated tumor arising from the upper pole of the right kidney that mimicked an adrenal gland or renal tumor, but later proved to be a SFT [4]. Yamamoto et al described a patient who underwent Tc-99m MIBI parathyroid scinitigraphy, which showed uptake highly suggestive of an ectopic parathyroid adenoma, but also later proved to be a SFT [5]. Our case appears to be the first documented case of malignant pleural SFT in an NF1 patient, which also showed signs of mimicry, as seen in the first two cases. Initial radiologic evaluation of the pleural mass suggested nerve sheath tumor, especially in the setting of other clinical findings of neurofibromatosis such as plexiform neurofibroma, as well as presence of a para-orbital mass. Nonetheless, other neoplastic causes such as pleural lipoma, pleural fibrosarcoma, mesothelioma, and metastatic pleural disease could not be ruled out by imaging alone without a definitive biopsy of the mass. Therefore, fine needle aspiration of the mass was performed and showed diagnostic features of SFT and subsequent excision showed areas of malignant transformation.
While NF1 patients have an increased risk of developing various tumors and malignancies, there is no definitive link between NF1 and SFT due to lack of published data [15]. Hence, it is difficult to comment on the exact relationship between these two disease processes. Whether or not NF1 is associated with the development of SFT and the relationship between such, as well as possible malignant transformation of SFT would need to be investigated further.
To Know More About Cancer Therapy & Oncology International Journal Please click on:
https://juniperpublishers.com/ctoij/index.php
To Know More About Open Access Journals Please click on:
https://juniperpublishers.com/index.php
#Juniper publishers#Open access Journals#Peer review journal#Juniper publisher reviews#Juniper publisher journals
0 notes
Text
Neurofibromatosis Treatment Drugs Market Forecast - 2027
To Gain More Insights into the Neurofibromatosis Treatment Drugs Market, Browse Summary of the Research Report –
Neurofibromatosis is a genetic disorder of the nervous system. It mainly affects growth of nerve cells. It is characterized by the growth of tumors on nerves. Neurofibromatosis is considered as a hereditary disease which mainly occurs in children or it can happen due to mutation (change) of genes. Usually the Neurofibromatosis tumors are benign, but sometimes they can become cancerous. There are three types of neurofibromatosis: Type 1 (NF1), Type 2 (NF2), and Schwannomatosis. All three types show varied symptoms.
https://www.coherentmarketinsights.com/market-insight/neurofibromatosis-treatment-drugs-market-4188
There are three types of Neurofibromatosis; Neurofibromatosis type 1, Neurofibromatosis type 2, and Schwannomatosis. Neurofibromatosis type (NF) 1 is a common monogenic disorder of neurocutaneous tissue growth that arises secondary to mutations in the tumor suppressor gene NF1. It features an autosomal dominant pattern of inheritance. Individuals with NF1 typically have an increased predisposition to a variety of benign and malignant tumors and develop neurofibromas, and axillary and inguinal freckling. Neurofibromatosis 2 (NF2) is less common than NF1. Common signs and symptoms of NF 2 include benign, slow-growing tumors affecting the cranial, spinal, and peripheral nerves, as well as the meninges. KOSELUGO- selumetinib is the only drug available for the treatment of Neurofibromatosis Type-1. Moreover, some drugs such as bevacizumab are also used as off-label for the treatment of Neurofibromatosis
Increasing focus of major players on research and development of novel therapies for treating neurofibromatosis. Moreover, clinical studies are well supported by regulatory authorities which is expected to drive the growth of the global neurofibromatosis treatment drugs market. For instance, in October 2019, SpringWorks Therapeutics, Inc., a clinical-stage biopharmaceutical company focused on developing life-changing medicines for patients with severe rare diseases and cancer, initiated the Phase 2b ReNeu clinical trial to evaluate the mirdametinib (formerly PD-0325901), an oral, small molecule designed to inhibit MEK1 and MEK2, in children and adult patients with neurofibromatosis type 1 (NF1)-associated plexiform neurofibromas (NF1-PN). In July 2019, the European Commission granted Orphan Drug Designation for SpringWorks Therapeutics’ mirdametinib (formerly PD-0325901), an oral, small molecule inhibitor of MEK1 and MEK2, for the treatment of neurofibromatosis type 1 (NF1).
“We Do Offer Sample of Neurofibromatosis Treatment Drugs Market Report. Kindly go through the follow information in order to access sample copy.”
https://www.coherentmarketinsights.com/insight/request-sample/4188
Table of Contents
Research Objectives and Assumptions
Research Objectives
Assumptions
Abbreviations
Market Purview
Report Description
Market Definition and Scope
Executive Summary
Market Snippet, By Drug Type
Market Snippet, By Distribution Channel
Market Snippet, By Region
Coherent Opportunity Map (COM)
Market Dynamics, Regulations, and Trends Analysis
Market Dynamics
Drivers
Restraints
Market Opportunities
Impact Analysis
Market Trends
Regulatory Scenario
Reimbursement Scenario
Epidemiology
PEST Analysis
Recent Product Launch
Merger and Acquisition Scenario
Top players in the market
Major Player operating in the global neurofibromatosis treatment drugs market is AstraZeneca Plc.
Research methodology adopted by Coherent Market Insights
Coherent Market Insights followsa comprehensive research methodology focused on providing the most precise market analysis. The company leverages a data triangulation model which helps company to gauge the market dynamics and provide accurate estimates. Key components of the research methodologies followed for all our market reports include:
Primary Research (Trade Surveys and Experts Interviews)
Desk Research
Proprietor Data Analytics Model
In addition to this, Coherent Market Insights has access to a wide range of the regional and global reputed paid data bases, which helps the company to figure out the regional and global market trends and dynamics. The company analyses the industry from the 360 Degree Perspective i.e. from the Supply Side and Demand Side which enables us to provide granular details of the entire ecosystem for each study. Finally, a Top-Down approach and Bottom-Up approach is followed to arrive at ultimate research findings.
Request A Sample Copy Neurofibromatosis Treatment Drugs Market Report Click here:
https://www.coherentmarketinsights.com/insight/request-sample/4188
Get PDF Research Report Brochure @
https://www.coherentmarketinsights.com/insight/request-pdf/4188
Buy Now this Premium Report to Grow your Business @
https://www.coherentmarketinsights.com/insight/buy-now/4188
About Us:
Coherent Market Insights is a global market intelligence and consulting organization focused on assisting our plethora of clients achieve transformational growth by helping them make critical business decisions.
Contact Us:
Name: Mr. Raj Shah
Email: [email protected]
Phone: US +12067016702
Country: United States
Visit our Blog: https://hospitalhealthcareblog.wordpress.com/
0 notes
Last Seen Blogs

dragneelshmagneel
Dragneel Shmagneel

haliibugg
so final jam is coming up


the-unseelie-court-official
Hello Humans

ossian94
Ladybird Wütend